超高效液相色谱串联质谱法测定2型糖尿病大鼠血浆阿托伐他汀和沙格列汀的浓度
2018-08-29汤道权肖冰心王添艳孙增先
董 洁 ,汤道权 ,肖冰心 ,王添艳 ,孙增先 △
(1.徐州医科大学附属连云港医院·连云港市第一人民医院,江苏 连云港 222000; 2.徐州医科大学,江苏 徐州 221004)
沙格列汀(SAX)为二肽基肽酶(DPP-4)抑制剂,单一或与其他降糖药联合使用,可控制患者血糖[1]。3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂(他汀类药物)可降低2型糖尿病(T2DM)患者心血管事件风险[2]。2013年版美国心脏病学院/美国心脏病协会(ACC /AHA)指南推荐[3],40 ~75 岁的 T2DM 患者需服用中等强度或高强度的他汀类药物。沙格列汀经肝脏代谢酶CYP3A4代谢生成5-羟基-沙格列汀(5-OH SAX)[4],两者都能选择性抑制 DPP -4 活性,增强其降糖效果。阿托伐他汀(ATO)在体内代谢也需要CYP3A4酶参与。目前尚未见报道沙格列汀及阿托伐他汀联合用药的相互作用,本研究中建立超高效液相色谱串联质谱法(UPLC-MS/MS)测定 ATO,SAX,5-OH SAX 的药物浓度,用于T2DM大鼠体内药代动力学研究。现报道如下。
1 仪器、试药与动物
仪器:Waters ACQUITY Sample Mange-FIN型液相色谱仪(美国 Waters公司);AB Qtrap 4 500型质谱仪,Analyst1.6.2数据处理软件(美国 ApplicedBiosystems公司);T1型氮气发生器(德国普利赛斯公司);AE-240型电子天平(瑞士梅特勒-托利多仪器有限公司);Peqlab多功能高速离心机(德国Deutschland公司);Vortex-genie 2漩涡混合器(美国ScientificIndustries公司)。
试药:阿托伐他汀钙(批号为20170320,纯度为99.9%,浙江海正药业股份有限公司);沙格列汀水合物对照品(批号为 SG-31204001,纯度为 94.36%,郑州华文化学品有限公司);5-羟基-沙格列汀(批号为20160303,纯度为 95.6%),维格列汀(批号为 20151203,纯度为99.5%),均由江苏豪森药业有限公司提供;乙腈(色谱纯,美国Tedia公司);醋酸铵(分析纯,国药集团化学试剂有限公司);Millipore超纯水。
实验动物:SD大鼠,雄性,SPF级,体质量80~120 g,购自上海西普尔-必凯实验动物有限公司,许可证号为SCXK(沪)2013-0016,饲养于徐州医科大学实验动物中心。所有动物实验均按徐州医科大学动物饲养和使用指南进行。
2 方法与结果
2.1 色谱条件与质谱条件
色谱柱:WatersBEHShieldRP18柱(100mm×2.1mm,1.7μm);流动相:10mmol/L 乙酸铵水溶液(A),乙腈(B),梯度洗脱,0~2.2 min,30% ~70%B,2.2~3.1 min,70% ~30%B,3.1 ~5.0 min,30%B;流速:0.2 mL /min;柱温:30 ℃;进样量:10 μL。
采用电喷雾电离源(ESI),以多反应监测(MRM)模式扫描,正离子方式检测。离子化电压:5 500 V;温度:350 ℃;喷雾气(N2)压力:60 psi;辅助加热气(N2)压力:50 psi;气帘气体(N2)压力:35 psi;碰撞气模式:Medium;扫描时间:100 ms。用于定量分析的离子反应 m/z分 别为 559.3→440.2(ATO),316.2→180.2(SAX),332.3→196.2(5 -OH SAX),304.2→153.9(维格列汀,内标)。
2.2 溶液制备与血浆样品处理
对照品溶液:取ATO,SAX,5-OH SAX对照品,精密称定,ATO用水-甲醇(50∶50)溶解并稀释,SAX及5-OH SAX用水-乙腈(50∶50)溶解并稀释,配成质量浓度皆为1 g/L的混合对照品贮备液。用水-甲醇(50∶50)将ATO及5-OH SAX依次稀释成质量浓度为10,25,50,100,250,500,1 000,2 500,5 000 ng /mL 的溶液,SAX 质量浓度为 20,50,100,200,500,1 000,2 000,5 000,10 000 ng/mL 的对照品工作液。
内标(IS)溶液:取维格列汀对照品,精密称定,用水-乙腈(50∶50)溶解并稀释,配成质量浓度为1 g/L的内标贮备液。临用前,用水-甲醇(50∶50)稀释定容至500 ng/mL。配置好的对照品贮备液及内标贮备液置-20℃冰箱贮存,备用。
混合血浆样品:取适量ATO,SAX,5-OH SAX标准工作液,加入空白血浆,配制成含ATO及5-OH SAX质量浓度为 1,2.5,5,10,25,50,100,250,500 ng /mL的溶液,SAX 质量浓度为 2,5,10,20,50,100,200,500,1 000 ng/mL的系列混合血浆样品溶液。同法制备定量下限、低、中、高质量浓度的质控血浆样品溶液,即ATO及 5 - OH SAX(1,2.5,50,250 ng/mL),SAX(2,5,100,500 ng/mL),-20℃贮存,备用。
血浆样品处理:取空白血浆50 μL,加入500 ng/mL内标工作液 10 μL、乙腈 250 μL,涡旋振荡 3 min,13 500 r/min离心10 min,移取上清液,在室温下氮气吹干。残留物用100 μL水-甲醇(50∶50)复溶,涡旋振荡1 min,13 500 r/min 离心 5 min,取上清液 10 μL,进样。
2.3 方法学考察
专属性考察:分别取6个SD大鼠空白血浆样品;空白血浆加入ATO,SAX,5-OH SAX及内标溶液;大鼠给药0.5 h后收集血浆样品,按照2.2项下血浆样品操作,行 UPLC-MS/MS 分析,色谱图见图 1。ATO,SAX,5-OH SAX 及 IS的保留时间分别为 3.77,1.91,1.63,1.67 min,保留时间 1.01 min 处发现内源性物质峰,但不干扰ATO,SAX,5-OH SAX及内标溶液的测定,为了尽量减少内源性物质对仪器的污染,将1.40 min之前的组分切外,结果见图1。
标准曲线制备及定量下限考察:取2.2项下混合血浆样品溶液,按血浆样品处理方法处理,并按拟订色谱及MS条件进样测定,记录色谱图。分别以待测物质量浓度(X)为横坐标、待测物峰面积与内标峰面积比值(Y)为纵坐标,用加权(W=1/X2)最小二乘法进行回归运算。ATO 标准曲线为 Y=0.00144X+0.00742(r=0.9986),定量限为1ng/mL;SAX标准曲线方程为 Y=0.00466X+0.002 12(r=0.999 5),定量下限为 2 ng/mL;5-OH SAX 标准曲线方程为 Y=0.009 07X+0.000 473(r=0.996 5),定量下限为 1 ng/mL。结果表明,ATO,SAX,5-OH SAX 质量浓度在 1~500,2~1 000,1~500 ng/mL范围内与峰面积线性关系良好。

1.ATO 2.SAX 3.5 - OH SAX 4.IS A.空白血浆 B.空白血浆加入ATO,SAX,5-OH SAX及IS C.SD大鼠灌胃ATO,SAX 0.5 h后的血浆样品加IS
精密度及准确度试验:分别制备含 ATO,SAX,5-OH SAX定量下限、低、中、高4个质量浓度的混合QC血浆样品溶液,各5份,按2.2项下方法处理,进样测定,并记录色谱图。连续测定3 d。采用当日所建立的标准曲线计算样品测定浓度,根据测定值与实际值进行计算,考察方法的精密度与准确度。结果显示,3 d内、日间 RSD<9.34%,准确度为92% ~110%,符合生物样品分析的要求。结果见表1。

表1 ATO,SAX,5-OH SAX日内及日间精密度与准确度实验结果
提取回收率和基质效应试验:取ATO,SAX,5-OH SAX混合QC血浆样品(n=5),按2.2项下方法操作,进样分析,记录相应分面积(A)。取空白血浆 50 μL,加入乙腈250 μL,涡旋,离心,吹干上清液,在残渣中加入5 μL 含 ATO,SAX,5-OH SAX 的混合标样溶液(浓度与相应的质控样品一致),10 μL 内标,溶液 100 μL水-甲醇(50∶50)涡旋混匀,离心,取上清液进样分析,记录各组分峰面积(B)。以纯水代替空白血浆,进行上述操作,进样分析,记录各组分峰面积(C)。基质效应=A/C×100%,提取回收率=A/B×100%。结果表明,ATO,SAX,5-OH SAX及内标的基质效应为 84% ~107%,提取回收率为89% ~103%(n=5),RSD<11%。表明在本试验所选择的色谱条件下,可忽略基质效应影响。
稳定性试验:分别考察混合质控血浆样品在不同保存条件下的稳定性(n=3)。血浆样品在室温下放置24 h,-80℃保存60 d及反复冻融3次等处理后,按2.2项下方法操作,进样分析。结果 ATO,SAX,5-OH SAX 的相对误差(RE)值分别为 0.7 ~10.6,0.8 ~8.7,2.1 ~ 10.1。
2.4 药代动力学研究
取6只SD大鼠,普通饲料适应性喂养1周,给以高脂饲料喂养4周,大鼠禁食、不禁饮12 h,给以链脲佐菌素(STZ,40 mg /kg)腹腔注射,诱导糖尿病模型,选择造模成功的大鼠模型进行实验。T2DM大鼠同时给予SAX及ATO各10 mg/kg灌胃。分别于给药前后0,0.25,0.5,0.75,1,1.5,2,3,4,6,8,12 h 时采血,各 0.5 mL,置EP管,血样在 4℃下,4 000 r/min离心 15 min,取上层血浆于-80℃冰箱保存。采用UPLC-MS/MS法测定不同时间点的ATO,SAX,5-OH SAX的血药浓度和药-时曲线,详见图2。采用Win Nolin 6.1软件按照非房室模型进行处理,计算药代动力学参数,结果见表2。

图2 T2DM大鼠体内ATO,SAX,5-OH SAX的药-时曲线
表2 ATO,SAX,5-OH SAX在T2DM大鼠体内的药代动力学参数± s)
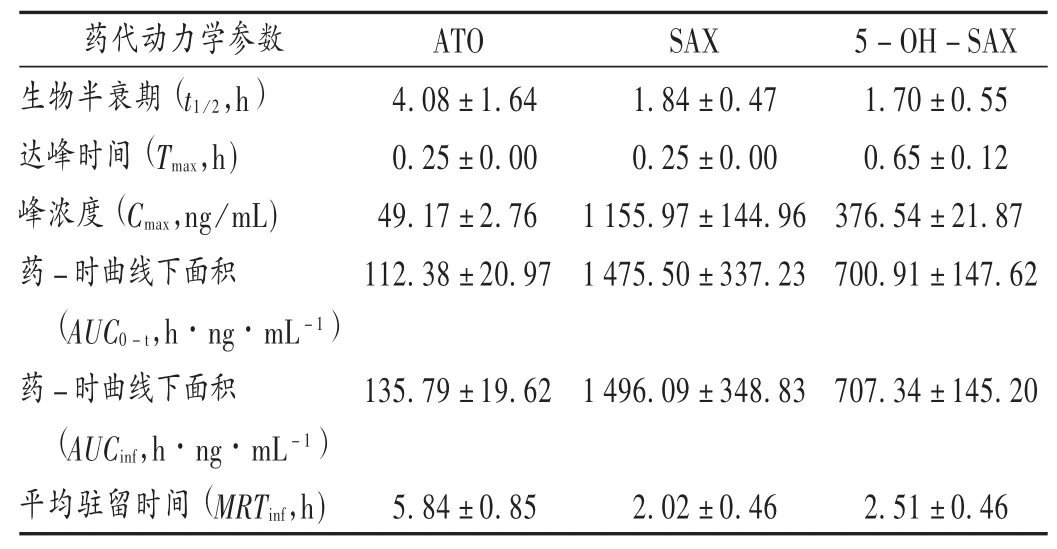
表2 ATO,SAX,5-OH SAX在T2DM大鼠体内的药代动力学参数± s)
药代动力学参数ATO SAX 5-OH-SAX生物半衰期(t1/2,h)达峰时间(Tmax,h)峰浓度(Cmax,ng/mL)药-时曲线下面积(AUC0-t,h·ng·mL-1)药-时曲线下面积(AUCinf,h·ng·mL-1)平均驻留时间(MRTinf,h)4.08 ± 1.64 0.25 ± 0.00 49.17 ± 2.76 112.38 ± 20.97 1.84 ± 0.47 0.25 ± 0.00 1 155.97 ± 144.96 1 475.50 ± 337.23 1.70 ±0.55 0.65 ± 0.12 376.54 ± 21.87 700.91 ± 147.62 135.79 ± 19.621 496.09 ± 348.83707.34 ± 145.20 5.84 ± 0.852.02 ± 0.462.51 ±0.46
3 讨论
T2DM患者需服用调脂药及降糖药,以预防心血管疾病风险,故可同时测定ATO及SAX药物浓度的方法对其药代动力学研究至关重要。目前,国内外与ATO及SAX血药浓度测定的方法有很多,可采用RP-HPLC/UV[5],LC - ESI- MS,GC - MS[6]及 HPTLC - UVD[7]等方法测定血浆中ATO浓度,SAX的测定主要采用GC[8],LC -MS /MS[9]等方法。相对于本研究中所描述的方法,均具有检测灵敏度不足、采集样本量大、出峰时间长等缺点,且不能同时测定血浆样品中ATO及SAX药物浓度。故对其作适当改进,增强检测灵敏度,样品保留时间缩短到5 min,提高了分析准确度、灵敏度及分析效率。
优化固定相时,曾尝试Acquity UPLC HSS T3 C18色谱柱、Waters BEH C18色谱柱及Waters shield RP C18色谱柱,结果发现,ATO在Acquity UPLC HSS T3 C18色谱柱无吸收峰,使用Waters BEH C18色谱柱,待测物都能检测出,但其中5-OH SAX的响应值过低,但这些问题在Waters shield RP C18色谱柱上均能改善,待测物具有很好的灵敏度及峰形。在流动相条件优化时,曾采用10 mmol/L乙酸铵-乙腈为流动相,开始时尝试等度洗脱,大水相条件下,SAX及其代谢物5-OH SAX能分开,但ATO不能很好地分离,随着有机相比例的增加,最终在有机相为70%时,ATO被很好地分离出来。故待测物在梯度洗脱条件下,有很好的响应值及峰形。
血浆样品前处理对于色谱方法的建立至关重要,将直接影响待测物的可靠性及准确性。通过查阅文献[10-11]发现,ATO及SAX都有采用乙腈沉淀蛋白的方法提取样品,由于SAX极性大,而ATO极性小,因而在吹干后的复溶采用50%的甲醇。结果表明,采用乙腈蛋白质沉淀法处理血浆样品,待测物的基质效应及提取回收率均符合规定[12]。
本研究中采用UPLC-MS/MS法同时测定T2DM大鼠灌胃给药后血浆中ATO,SAX及5-OH SAX的含量,以维格列汀为内标,在正离子模式下检测。结果表明,此方法具有良好的精密度与准确度,符合生物样品分析方法要求,灵敏度和专属性高。研究了ATO联合SAX在T2DM大鼠体内的药代动力学过程,确定了本研究中所建立的方法在非临床药代动力学研究中的适用性,可为其非临床药代动力学及毒理学等研究提供分析方法。
