磷酸三丁酯和磷酸二丁酯与Pu(Ⅳ)配合物的密度泛函研究
2016-11-11吕洪彬晏太红
吕洪彬,左 臣,晏太红
中国原子能科学研究院 放射化学研究所,北京 102413
磷酸三丁酯和磷酸二丁酯与Pu(Ⅳ)配合物的密度泛函研究
吕洪彬,左臣,晏太红
中国原子能科学研究院 放射化学研究所,北京102413
采用B3LYP密度泛函方法,Pu(Ⅳ)和其他原子分别采用相对论有效原子实势(RECP)和6-311 g(d,p)全电子基组,研究了Pu(NO3)4TBP2和Pu(NO3)2DBP2的几何结构、热力学参数及紫外可见光谱。几何结构计算结果表明,由于配体空间位阻的影响,Pu与DBP的键长更短,自然布居分析(NPA)结果也表明,Pu与DBP之间的电荷转移更多,因此二者之间相互作用更强。热力学分析显示,Pu(NO3)4TBP2与HDBP发生配体交换的反应自由能为-178.9 kJ/mol,说明DBP与Pu之间的相互作用更强。Pu(NO3)4TBP2的紫外吸收光谱主要来源于硝酸根到Pu的f轨道的跃迁,TBP贡献不大;而Pu(NO3)2DBP2的跃迁同时来源于硝酸根和DBP到Pu的f轨道的配体-金属电荷转移跃迁(LMCT)。
钚;磷酸三丁酯(TBP);磷酸二丁酯(HDBP);密度泛函理论
利用磷酸三丁酯(TBP)作为萃取剂的普雷克斯(PUREX)流程是核燃料后处理中最为成熟的技术。在萃取过程中TBP和稀释剂都会在辐射条件下发生辐解,产生包括磷酸二丁酯(HDBP)在内的辐解产物[1-2]。在实验和工厂运行中存在Pu(Ⅳ)不能被充分反萃的现象,而有机相中TBP的辐解产物(HDBP)与Pu(Ⅳ)的结合被认为是使其无法被充分反萃的重要因素之一[3]。对TBP和HDBP与Pu(Ⅳ)的配合物结构和性质的研究将会有助于萃取过程的机理研究和Pu(Ⅳ)回收率的提高。已有文献[4-6]在实验上对Pu(Ⅳ)和TBP、HDBP形成的配合物组成、平衡常数等性质进行了研究,但由于Pu的毒性与放射性,对其结构的研究比较少。对钚配合物的理论研究既是相对匮乏的实验数据的补充,还能够加深对钚配合物的认识。近年来,采用B3LYP、PBE0等泛函的密度泛函理论(DFT)方法被应用于锕系元素配合物的结构、性质研究及核燃料后处理流程相关的研究中[7-10]。相对论效应、溶剂效应等因素的引入使得理论计算的结果精确度明显提高。本工作拟在已有实验基础上,采取DFT方法对Pu(Ⅳ)和TBP、HDBP形成的配合物结构进行研究,并对其热力学参数进行计算,同时对其紫外光谱进行考察和分析,这些结果将有助于对有机相中TBP和HDBP的Pu(Ⅳ)配位性质的理解,同时为工艺改进提供理论基础。
1 方法和基组
所有的计算均采用密度泛函理论,利用Gaussian 09软件包[11],选取B3LYP杂化泛函进行不考虑自旋-轨道耦合的标量相对论计算。对钚原子的计算考虑相对论效应,对内层电子采用相对论有效原子实势(RECP),对Pu(Ⅳ)配合物采用五重态自旋态,对U(Ⅳ)配合物采取单重态自旋态。对钚原子采用相对论有效原子实势基组,在进行结构计算时,对钚原子选取德国斯图加特大学ECP78MWB_SEG 基组进行计算[12],这个基组包含78个中心电子和16个价电子,在进行自由能及紫外光谱计算时,采取德国斯图加特大学ECP60MWB_SEG 基组,这个基组包含60个中心电子和34个价电子。本次计算中没有考虑自旋-轨道耦合效应。对其他原子采取6-311g(d,p)全电子基组[13],有研究结果表明,采用B3LYP/RECP/6-311g(d,p)的理论计算方法对锕系元素及其与有机配体形成的配合物进行计算得到的结果是可靠的[9]。所有的结构优化都是在气相环境中,采用B3LYP/RECP/6-311g(d,p)的理论计算方法进行的。在优化结构的基础上,采用相同的方法对振动频率进行计算,在气象环境中得到配合物的自由能以及零点能。采用Multiwfn程序对自然原子电荷和Mayer轨道键级进行分析。在气相环境优化结构的基础上,利用似导体屏蔽模型得到有机溶剂环境下的自由能以及零点能。利用含时-密度泛函方法(TD-DFT)对其紫外光谱进行模拟同时对紫外峰对应的跃迁进行了分析。为节省计算量,将TBP及HDBP中的丁基替换为甲基。去质子化的HDBP用DBP表示。
2 计算结果
2.1Pu(NO3)4TBP2和Pu(NO3)2DBP2的几何构型
依据文献[4]中钚的TBP和DBP配合物组成,优化得到了气相环境中Pu(NO3)4TBP2和Pu(NO3)2DBP2两种化合物的结构,示于图1。

Pu(NO3)4TBP2和Pu(NO3)2DBP2的结构参数与自然布居分析(NPA)结果列入表1。NPA分析显示,Pu(NO3)4TBP2中Pu电荷为1.863,Pu(NO3)2DBP2中Pu电荷为1.601,说明DBP与Pu原子之间电荷转移更多,这导致了DBP与Pu的结合能比TBP与Pu的结合能更大。计算表明,去离子化的HDBP中的配位氧原子电荷(-1.116)比TBP中的配位氧原子电荷(-1.074)更多,说明DBP中氧原子的配位能力比TBP中氧原子的配位能力更强。
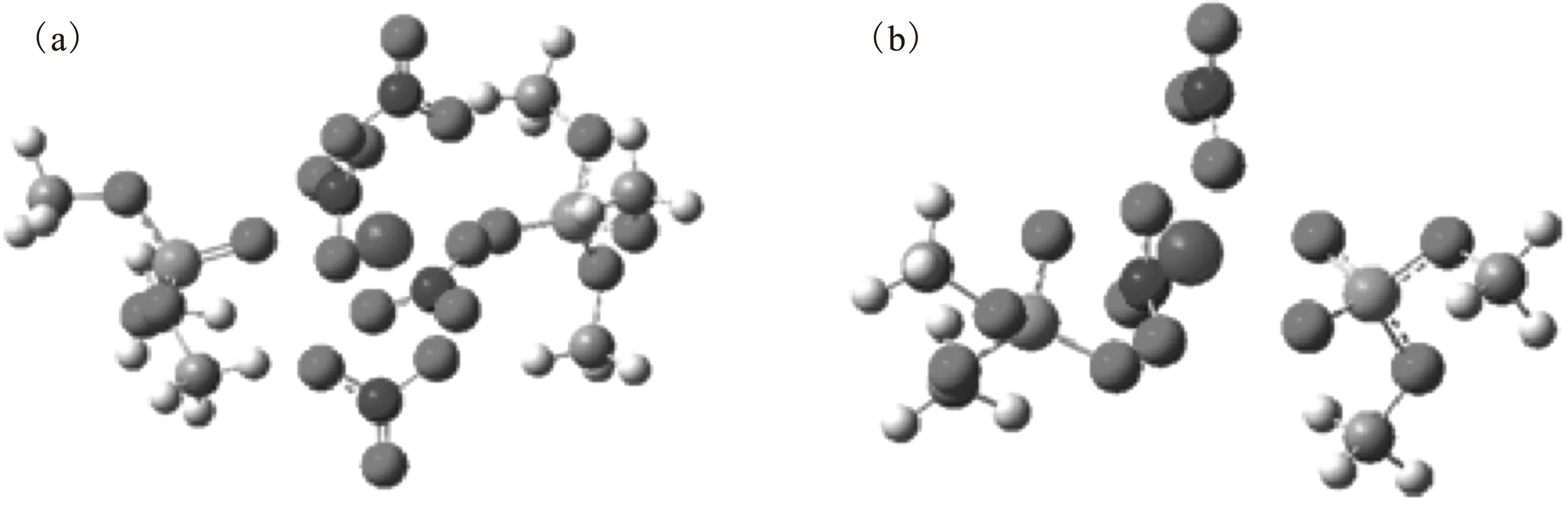
浅蓝色代表Pu,红色代表O,蓝色代表N,橙色代表P,灰色代表C,白色代表H图1 Pu(NO3)4TBP2(a)和Pu(NO3)2DBP2(b)的结构Fig.1 Optimized structure of Pu(NO3)4TBP2(a) and Pu(NO3)2DBP2(b)
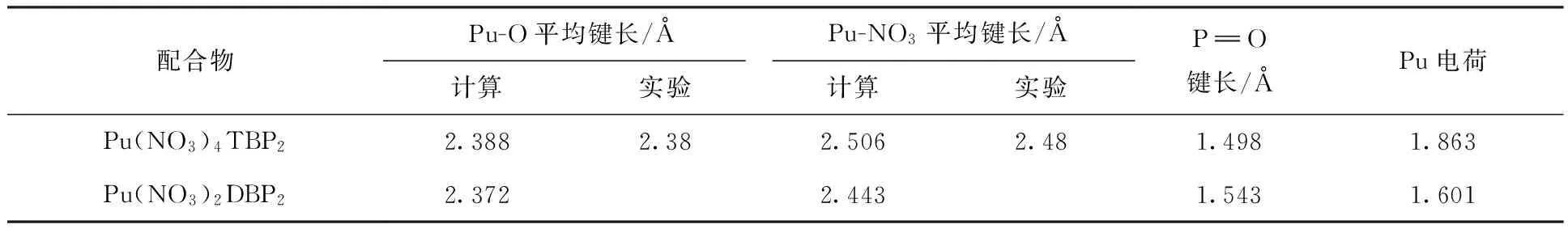
表1 Pu(NO3)4TBP2和Pu(NO3)2DBP2的结构参数与NPA分析结果Table 1 Structure parameters and NPA analyze results of Pu(NO3)4TBP2 and Pu(NO3)2DBP2
对配合物中Pu-O的Mayer键级进行了分析[15],Mayer键级越高,化学键的共价成分就越多。在Pu(NO3)4TBP2中Pu-OTBP的Mayer键级为0.28左右,Pu-ONO3的键级为0.27~0.43,在Pu(NO3)2DBP2中Pu-ODBP的Mayer键级为0.52~0.55,Pu-ONO3的键级为0.40~0.51,这些结果说明Pu与O之间有一定的共价成分。Pu-OTBP的Mayer键级比Pu-OTBP的键级高,说明二者之间的共价成分更多,因此,Pu与DBP的相互作用更强。Pu(NO3)2DBP2中Pu-ONO3的键级比Pu(NO3)4TBP2中Pu-ONO3的键级更高,说明Pu(NO3)2DBP2中Pu-ONO3的作用更强,这主要是由于空阻原因导致Pu(NO3)2DBP2中Pu-ONO3的距离更短,使得二者相互作用更强。与自由Pu(NO3)4中Pu-ONO3的键级(0.49~0.54)相比,TBP和DBP与Pu配位后,Pu-ONO3的键级都有减少,Pu(NO3)4TBP2中减少的更加明显。
2.2Pu(NO3)4TBP2和Pu(NO3)2DBP2的热力学性质研究
实验结果表明,在有机相环境中,DBP与Pu(Ⅳ)配合物的稳定常数比TBP与Pu(Ⅳ)的稳定常数大,说明DBP与Pu(Ⅳ)的相互作用更强。为了研究TBP和DBP对Pu(Ⅳ)的热力学选择性,首先在气相条件下对反应(1)的热力学参数进行了计算,结果列入表2。由表2可知:ΔH=20.36 kJ/mol;ΔS=44.40 J/K,说明该反应是熵驱动的反应。配体交换过程是配体解离,新配体再重新配位的过程。配体解离导致熵增,配体配位导致熵减,反应过程中伴随着TBP和DBP交换的过程有两个硝酸根从配合物中解离下来,因此导致整个反应的熵增加。虽然反应熵值为正值,但是由于焓变的正值较大,所以计算得到的反应自由能ΔG=7.13 kJ/mol,因此在这种条件下,配位反应难以发生。
Pu(NO3)2DBP2+2TBPHNO3
(1)
ΔG=ΔG(Pu (NO3)2DBP2)+
2ΔG(TBPHNO3)-ΔG(Pu(NO3)4TBP2)-
2ΔG(HDBP)
(2)
虽然气相计算结果表明,TBP与Pu(Ⅳ)的结合能力更强,为使计算结果与实际状态更加接近,因此在考虑溶剂情况下进行了计算。因为Pu萃取实验的有机相通常为煤油或正十二烷,因此采用正己烷作为模拟溶剂模型,计算结果列入表2。由表2可知:与在气相条件中计算结果不同,在溶剂条件下计算得到反应(1)的ΔH=-154.97kJ/mol,在溶剂条件下反应焓变转为负;ΔS=80.27J/K,在溶剂条件下熵变增大,更有利于反应的进行,由此计算得到的反应自由能ΔG=-178.89kJ/mol,在液相中反应转变为热力学有利的反应。热力学计算结果说明,DBP能够和Pu(NO3)4TBP2配位,说明与TBP相比,DBP与Pu(Ⅳ)的配位能力更强。
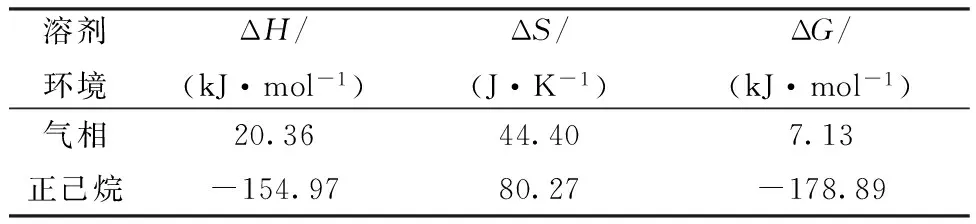
表2 利用B3LYP/6-311G(d,p) 计算得到的反应(1)的热力学参数Table 2 Calculated thermodynamic parameters of reaction (1) by B3LYP/6-311G(d,p) methods
2.3Pu(NO3)4TBP2和Pu(NO3)2DBP2吸收光谱的理论研究
利用TD-DFT方法对气相中Pu(NO3)4TBP2和Pu(NO3)2DBP2的紫外光谱进行了模拟,分子轨道示于图2、3。Pu(NO3)4TBP2的吸收峰主要为377、407、581、607 nm,分别对应HOMO-2到LUMO+2、HOMO-3到LUMO、HOMO-2到LUMO和HOMO-1到LUMO的跃迁。由图2可知,Pu(NO3)4TBP2的HOMO-3、HOMO-2、HOMO-1和HOMO轨道其主要由硝酸根中的N、O原子的p轨道构成,LUMO轨道主要由Pu原子f轨道组成,因此其紫外光谱主要对应于硝酸根中的N、O原子的p轨道到Pu原子f轨道的配体-金属电荷转移跃迁(LMCT),TBP分子的轨道对紫外光谱跃迁贡献不大。

图2 气相中Pu(NO3)4TBP2的分子轨道Fig.2 Molecular orbitals of Pu(NO3)4TBP2 in gas phase
Pu(NO3)2DBP2的吸收峰主要在405、420、432、487、513、612、630 nm,分别对应于HOMO-4到LUMO+2、HOMO-2/HOMO-1到LUMO+2、HOMO-6到LUMO/HOMO-3到LUMO+1、HOMO-2/HOMO-4到LUMO+1、HOMO到LUMO+1、HOMO到LUMO的跃迁。由图3可知,HOMO-6、HOMO-4、HOMO-3、HOMO-1主要由DBP中原子轨道组成,HOMO-2和HOMO主要由硝酸根中原子轨道组成,LUMO、LUMO+1和LUMO+2主要由Pu的f轨道组成,因此405、420、32 nm处吸收峰主要对应于DBP到Pu的配体-金属电荷转移跃迁(LMCT),487 nm吸收峰由DBP到Pu的LMCT和硝酸根到钚的LMCT混合而成,513、612、630 nm则主要是硝酸根到钚的LMCT。

图3 气相中Pu(NO3)2DBP2的分子轨道Fig.3 Molecular orbitals of Pu(NO3)2DBP2 in gas phase
3 结 论
利用B3LYP密度泛函理论方法,采用Gaussian 09程序和相对论赝势基组,对Pu(NO3)4TBP2和Pu(NO3)2DBP2两种化合物进行了结构优化。结果表明:Pu(NO3)4TBP2的优化结构与实验结果相吻合,Pu(NO3)2DBP2的优化结构中,Pu与DBP和硝酸根的键长都比Pu(NO3)4TBP2中Pu与TBP和硝酸根的键长要短,相互作用更强。在优化结构基础上进行NPA分析,发现在Pu(NO3)2DBP2中,DBP到Pu的电荷转移比Pu(NO3)4TBP2中TBP与Pu的电荷转移更多,且DBP与Pu之间的Mayer键级比TBP与Pu的键级更大。利用TD-DFT方法对紫外可见光谱进行了模拟并对吸收峰所对应的轨道跃迁进行了分析。对紫外光谱的比较可以发现,Pu(NO3)2DBP2在400~430 nm处的紫外吸收峰更强,这一部分主要对应于DBP到Pu的LMCT跃迁,而在Pu(NO3)4TBP2中,TBP分子则对紫外吸收贡献较少。
[1]Mincher B J, Elias G, Martin L R, et al. Radiation chemistry and the nuclear fuel cycle[J]. J Radioanal Nucl Chem, 2009, 282(2): 645-649.
[2]Bellido A V, Rubenich M N. Influence of the diluents on the radiolytic degradation of TBP in TBP, 30% (V/V)-diluent-HNO3systems[J]. Radiochim Acta, 1984, 36: 61-64.
[3]Mincher B J, Mezyk S P. Radiation chemical effects on radiochemistry: a review of examples important to nuclear power[J]. Radiochim Acta, 2009, 97(9): 519-534.
[4]May I, Taylor R J, Wallwork A L, et al. The influence of dibutylphosphate on actinide extraction by 30% tributylphosphate[J]. Radiochim Acta, 2000, 88(5/2000): 283-290.
[5]Sokhina L P, Solovkin A S, Teterin E G, et al. Dibutylphosphates of tetravalent plutonium, zirconium and thorium, formed in solutions of tri-n-butyl phosphate (TBP)[J]. Radiokhimiva, 1978, 20(1): 28-31.
[6]Sulka M, Cantrel L, Vallet V. Theoretical study of plutonium(Ⅳ) complexes formed within the PUREX process: a proposal of a plutonium surrogate in fire conditions[J]. J Phys Chem A, 2014, 118(43): 10073-10080.
[7]王东琪,van Gunsteren W F.锕系计算化学进展[J].化学进展,2011,23(7):1566-1581.
[8]Steele H, Taylor R J. A theoretical study of the inner-sphere disproportionation reaction mechanism of the pentavalent actinyl ions[J]. Inorg Chem, 2007, 46(16): 6311-6318.
[9]Xiao C L, Wu Q Y, Wang C Z, et al. Quantum chemistry study of uranium(Ⅵ), neptunium(Ⅴ), and plutonium(Ⅳ,Ⅵ) complexes with preorganized tetradentate phenanthrolineamide ligands[J]. Inorg Chem, 2014, 53(20): 10846-10853.
[11]Frisch M J,Trucks G W,Schlegel H B,et al. Gaussian 09, Revision A.1[R]. U S A: Gaussian, Inc. Wallingford, CT, 2009.
[12]Kuchile W, Dolg M, Stoll H, et al. Energy-adjusted pseudopotentials for the actinides. parameter sets and test calculations for thorium and thorium molecules[J]. J Chem Phys, 1994, 100: 7535-7542.
[13]Dithchfield R, Hehre W J, Pople J A. Self-consistent molecular orbital methods extended gaussian-type basis for molecular-orbital studies of organic molecules[J]. J Chem Phys, 1971, 54: 724-728.
[14]Den Auwer C, Revel R, Charbonnel M C, et al. Actinide coordination sphere in various U, Np and Pu nitrato coordination complexes[J]. J Synchrotron Radiat, 1999, 6: 101-104.
[15]Lu T, Chen F W. Multiwfn: a multifunctional wavefunction analyzer[J]. J Comput Chem, 2012, 33(5): 580-592.
Density Functional Theoretical Analysis of Complexes of Tributylphosphate and Dibutylphosphate of Pu(Ⅳ)
LU Hong-bin, ZUO Chen, YAN Tai-hong
China Institute of Atomic Energy, P. O. Box 275(26), Beijing 102413, China
The geometry, thermodynamic selectivity and UV-vis spectra of Pu(NO3)4TBP2and Pu(NO3)2DBP2in the gas phase were investigated by using B3LYP density functional theory method with the RECP basis for Pu and 6-311 g(d,p) basis for other atoms. The structure optimization results show the bond length between Pu and DBP is shorter than the length between Pu and TBP for the steric hinder of the ligands, and NPA charge analysis shows more electron trasnfer between DBP and Pu ion indicating the stronger coordination ability of DBP. The free energy of ligand exchange between Pu(NO3)4TBP2and HDBP was -178.9 kJ/mol, indicating the higher coordination affinity between DBP and Pu. The UV-vis absorption spectra of Pu(NO3)4TBP2in the gas phase were pmainly from nitrate to f orbital of Pu, TBP ligand made few contribution, yet the absorption spectrum of Pu(NO3)2DBP2were both from nitrate to f orbital of Pu and the ligand-to-metal charge transfer between DBP and Pu.
plutonium; TBP; HDBP; density functional theory
2016-03-03;
2016-09-07
O634.12
A
0253-9950(2016)05-0282-06
10.7538/hhx.2016.38.05.0282
