大鼠软骨生成过程表达的小分子RNA生物信息学分析
2015-02-18邵世滨闵自信郭源旭王权成孙梦瑶
邵世滨,闵自信,郭源旭,王权成,孙梦瑶,韩 燕,孙 健
(1.西安交通大学医学部生物化学与分子生物学系,陕西西安 710061;2.山东医学高等专科学校生物化学教研室,山东临沂 276002)
非编码 RNA(non-coding RNA,ncRNA)是基因组转录出的一类不同于mRNA的遗传信息分子,包括小分子RNA(small RNA,sRNA)和长链非编码 RNA(long non-coding RNA,lncRNA),广泛地存在于各种生物中,而且随着生物复杂度的升高,基因组中的非编码序列的比例也相应增大,提示ncRNA在生物进化过程中的重要意义[1]。对真核细胞中ncRNA及其基因的发掘和功能研究,有可能揭示一个由ncRNA介导的遗传信息传递方式和表达调控网络,从不同于蛋白质编码基因的角度注释和阐明基因组的结构与功能,深入阐明生命活动的本质和规律[2]。
sRNA一般长度小于 200nt(nucleotide),由RNA聚合酶Ⅱ或RNA聚合酶Ⅲ所合成,种类众多,通常与蛋白质组成复合物,在细胞的生命活动中起重要的作用[3]。按照细胞内空间定位,sRNA分为细胞核小RNA(small nuclear RNA)和细胞质小scRNA(small cytoplasmic RNA)两类。其中,研究最多是microRNA(miRNA,miR),长约20~24nt一类负调控因子,起源于RNA聚合酶Ⅱ转录的内源性单链sRNA[4]。其发现和深入研究进一步揭示了基因表达调控的复杂性,功能几乎包括了整个生物发育生长的过程,并不断发现新的功能[5]。
由于ncRNA没有编码蛋白质的读码框架,使得发现和鉴定sRNA有一定难度,常用方法有直接克隆测序技术、生物信息学预测、正向遗传学方法。第二代高通量测序技术可以一次性产出百万级的高质量sRNA序列,极大地弥补原来的技术缺陷,成为目前最有效、最常用的sRNA鉴定方法,尤其对于特定时空的细胞表达出的全部ncRNA谱的研究,是阐明其参与的调控网络的有效途径[6]。
针对sRNA的高通量测序的Illumina的Solexa平台,利用单分子阵列测试基因分型[7],通过大量的平行测序,深度鉴定并定量出全基因组水平的sRNA谱,而且可减少因被检测的核苷酸自身二级结构,导致的一段区域缺失。但是,高通量测序数据会出现大量假阳性、假阴性及其他的杂质(noise)。必须运用生物信息学和统计学结合的方法,对这些数据进行去规范化和标准化。
为了研究sRNA对骨骼发育与骨形成的调控作用,我们选择了软骨生成不同阶段的重要转折点:出生后d0(新生)、d21(断乳)、d42(性成熟)3个时间点的雌性SD大鼠股骨头软骨作为研究对象[8-9],使用Solexa测序平台鉴定了大于18nt的高质量sRNA序列表达,依次为:14 813 676、17 054 400、17 025 148条,并作生物信息学分析。
1 材料与方法
1.1 主要试剂、仪器及分析平台
1.1.1 主要试剂和仪器 Trizol®试剂(Invitrogen公司);DEPC(Amresco公司);Agarose(Solarbio公司);Tris base(Solarbio公司);RNA Marker RL6000(TaKaRa 公司);RevertAidTM First Strand cDNA Synthesis Kit(Fermentas,MBI公司);SYBR®Premix Ex TaqTMⅡ(TaKaRa 公司);Primers(Invitrogen公司)。Solexa Genome AnalyzerⅡ测序仪(Illumina公司)。
1.1.2 主要软件 ①SOAP(short oligonucleotide alignment program)[10];②MIREAP(https://sourceforge.net/projects/mireap/)。
1.1.3 数据库 ①SD大鼠基因组[11];②miRBase20.0 database;③Rfam9.1database。
1.2 实验动物及样本 实验选用的SPF级SD大鼠购于第四军医大学实验动物中心。动物在SPF级动物房繁育,环境维持室温(25±3)℃,相对湿度40%~60%,每日人工照明12h。动物垫料经高压蒸汽灭菌,动物饮灭菌凉开水,喂经钴60照射灭菌的颗粒饲料,自由进食及饮水。分别取d0,d21,d42龄远交系SD雌性大鼠各5只,断颈处死后,立即截取右后肢股骨,迅速剥下股骨头软骨组织,用冰冻的DEPC-生理盐水冲洗后,立即用Trizol®法提取总RNA,定量后将同一时间点的5个样本等量合并,每份样本都含有总RNA 20μg。
1.3 小分子RNA文库的构建及测序 ①使用聚丙烯酰胺凝胶电泳(PAGE)从20μg总RNA样本分离纯化出小于30nt的sRNA分子。②将分离出的sRNA分子与Solexa接头分子连接作为合成cDNA的模板,与接头分子互补的引物,进行12个PCR循环,获得长度约90bp的cDNA片段。③琼脂糖凝胶电泳中分离纯化cDNA扩增产物。④使用Illumina Genome Analyzer(Illumina,San Diego,CA,USA)对纯化的cDNA扩增产物进行序列分析,得到的所有数据直接被处理为高质量的图像文件。⑤去除接头序列和污染序列后,读取计算分析处理清洁序列[12]。
1.4 In silico分析策略 不同的样本由于测序深度等原因,其总量并不一样,不能直接对不同文库的测序拷贝数的结果进行比较。因此,须先进行“TPM标准化(normalize)”,即某种特定转录本的表达量用TPM(transcript per million,TPM)来表示(指每一百万的总转录本中,该转录本的拷贝数)。再进行:①重复序列的合并:为了减少计算量,分别将3个文库中的全部清洁序列sRNA完全相同序列合并,并计算拷贝数,获得unique sRNA序列。②TPM(transcript per million,TPM)标准化处理3个文库中每个unique sRNA的表达量。③SOAP:使用SOAP将unique sRNA序列与SD大鼠基因组比对,保留与基因组完全匹配(perfect match)的序列作进一步分析。④同源比对:将与基因组完全匹配的unique sRNA序列与miRBase20.0数据库比对,确定3个文库中表达出的已知miRNA。⑤剔除已知non-coding sRNA:将剩余数据与RNA数据库Rfam9.1比对,Rfam database包括除miRNA外的全部已知tRNA、rRNA、snRNA、snoRNA 和 其 他 non-coding RNA序列信息。剔除与Rfam database序列信息完全匹配的unique sRNA序列。
2 结 果
2.1 各样本小分子RNA文库表达的基本情况 使用高通量的Solexa平台测序技术,从d0、d21和d42三个大鼠软骨生成不同阶段的股骨头软骨细胞sRNA文库中,共读取到长度18nt至30nt的sRNA拷贝数,分别为:16 641 832条、18 560 892条和19 298 789条。为了简化测序数据,分别对3个文库读取到的所有sRNA进行分组和归类,序列完全相同的sRNA标签为同一序列,并统计其拷贝数。在剔除了低质量测序结果和接头序列,并SD大鼠基因组比对后,文库中包含了各自所表达的全部清洁序列sRNA[13],其中,d0包含有11 169 201条sRNA,d21包含有13 814 641条sRNA,d42包含有15 792 490条(表1)。
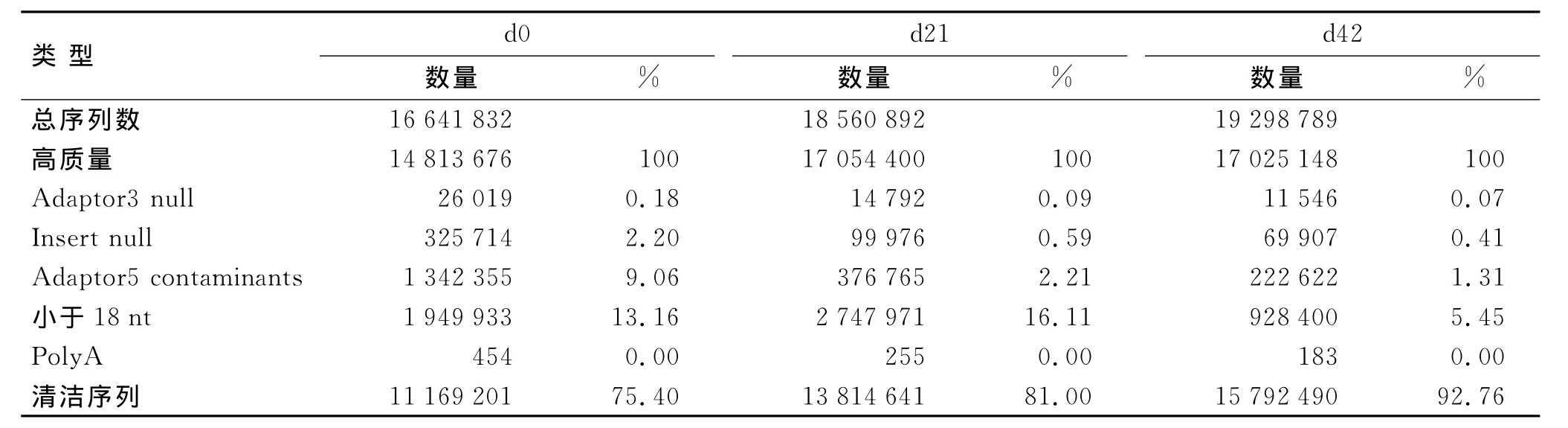
表1 sRNA测序的基本结果Tab.1 The summary of data produced by small RNA sequencing
统计d0、d21、d42的3个文库中,所有检测到的全部清洁序列sRNA长度,20~24nt的sRNA占比分别为71.94%、72.85%、86.39%;21~23nt的sRNA 占比分别为58.67%、60.17%、74.69%;22nt的sRNA占比分别为:29.71、28.19、37.76%,峰值均为22nt(图1A)。
2.2 In silico分析结果 ①各文库small RNA的重复序列分析:3个文库中的全部清洁序列sRNA中完全相同序列合并,并计算拷贝数,获得unique sRNA序列见表2。②各文库sRNA的基因组定位分析见表3。③各文库sRNA表达的miRNA:3个文库中,sRNA的长度分布峰值均为22nt,清洁序列的占比分别高达49.9%、47.2%,55.5%(图1B);成熟 miRNA的种类在unique sRNA种类中,占比分别为0.69%、0.78%和0.63%(表2)。④已知的ncRNA保留的与大鼠基因组完全匹配的sRNA序列,按照其生物起源和注释,分为rRNA、tRNA、snRNA、snoRNA、exon、intron等(表2)。⑤未知的ncRNA:剔除已知miRNA、miRNA*以及其它已知non-coding RNA后,剩余的无法与miRBase20.0和Rfam9.1匹配的未知sRNA,命名为“unann”。在d0、d21、d42文库中,unann占unique sRNA序列标签的比例,分别为54.24%、57.04%、54.09%,序列拷贝数分别是2 432 498、3 322 033、4 179 434(表2)。
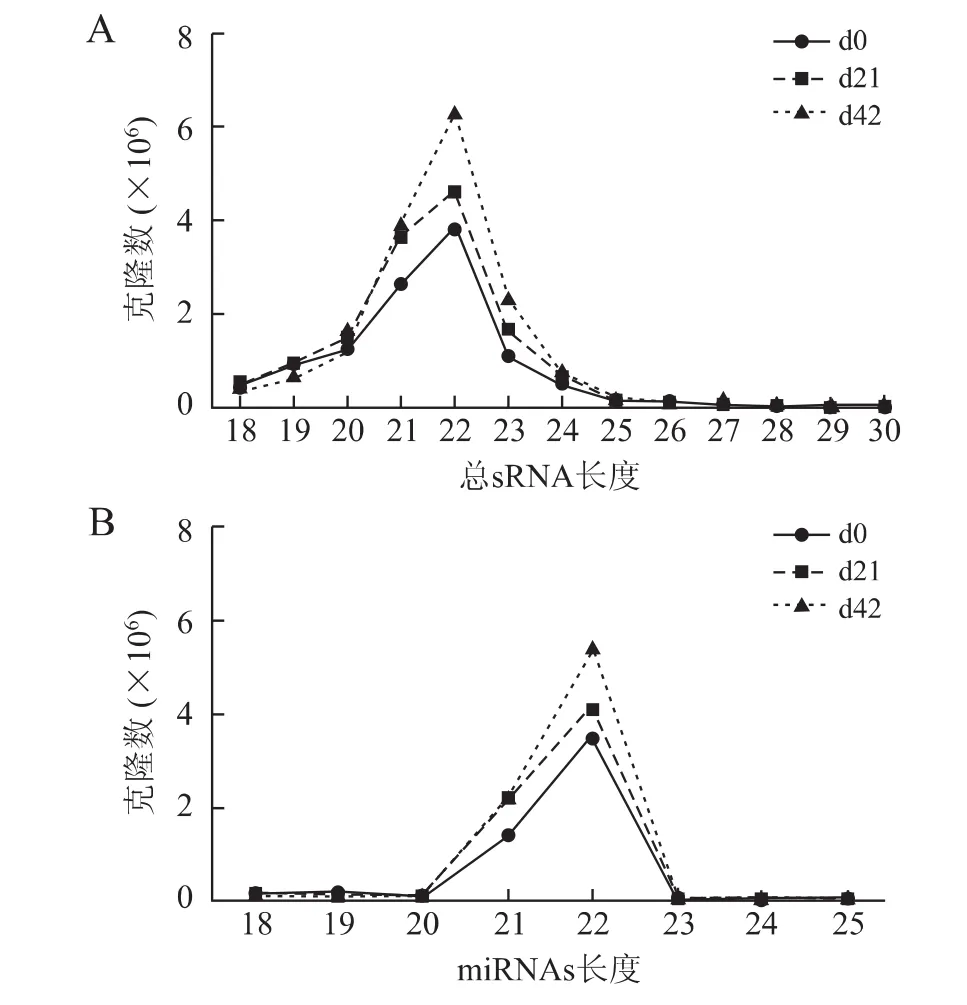
图1 3个关节软骨文库中mRNA的长度特征Fig.1 The length characteristics of rat articular cartilage miRNAs at different development stages
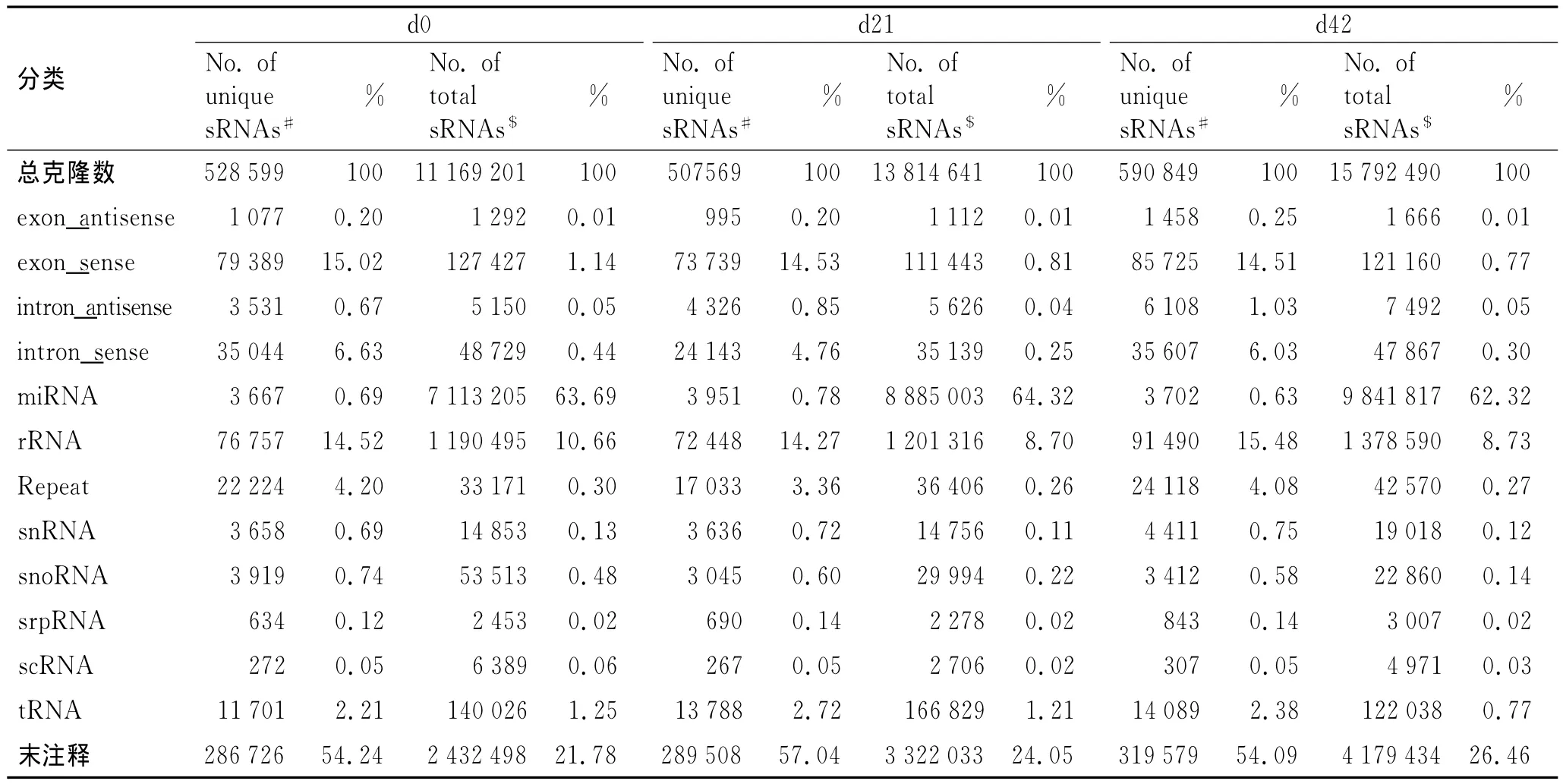
表2 大鼠软骨细胞中表达的sRNA数据统计Tab.2 The distribution of small RNAs in rat cartilage cells
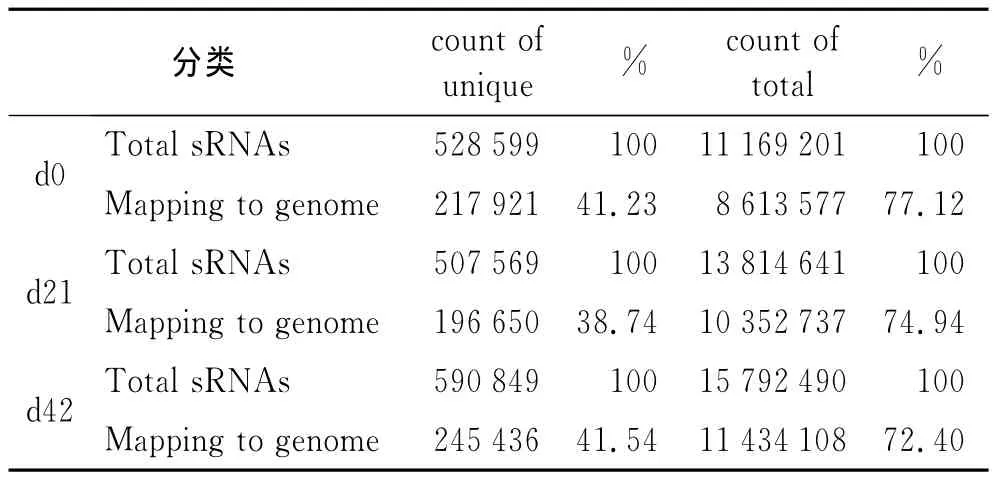
表3 sRNA基因组定位分析Tab.3 Mapping statistics of sRNA
3 讨 论
高通量测序技术进行sRNA测序,能够从基因组的角度研究sRNA的分布以及其在基因组上的染色质修饰情况,是目前发现特定发育阶段的sRNA最有效的方法,也是分析比较野生型和突变体中sRNA合成途径重要基因表达差异与功能的有效手段,还能够发现nat-siRNA和nat-miRNA等特殊作用方式的sRNA[14]。同时,避免了一代测序所固有的细菌克隆步骤,导致很小片段序列和极低丰度序列在克隆过程中的丢失现象,并能够精确的读取表达丰度极低的sRNA的拷贝数[15]。而且极大的提高了sRNA的发现效率,但是也产生了大量未知的候选sRNA的鉴定工作[15]。
大鼠从出生到性成熟,有着非常简短而迅速的成长过程。仅仅6周时间,大鼠就历经了婴幼儿期、儿童期和青春期这3个出生后重要的发育阶段。这一过程正是骨骼发育与骨形成、软骨细胞增殖与分化的重要过程[16]。因此,本研究中,我们以大鼠骨骼发育与骨形成的d0、d21、d42三个关键时点的股骨头软骨组织来构建sRNA文库。
但是,单只成年大鼠关节软骨组织非常有限,软骨组织中细胞比例也较低,RNA含量也较低,从软骨中提取高质量总RNA的难度相应也较高,sRNA占总RNA比例又低。因此,从一只大鼠股骨头软骨能够提取的总RNA量,不能达到Solexa测序构建sRNA文库所要求的单个样本总RNA量20μg的低限。所以,我们只能将同一时间点的5个雌性SD大鼠样本等量合并。
从3个文库序列分布情况来看,基本为20~24nt的序列,大部分为21~23nt,峰值为22nt的基本特征,完全符合高质量sRNA文库的特征。超过总拷贝数62%的清洁序列来自于成熟miRNA序列。一般认为,21nt和22nt miRNA倾向于与Ago蛋白构成沉默复合体,前者使靶基因转录本裂解,后者使靶基因转录本翻译阻遏;24nt miRNA的功能倾向于甲基化组蛋白,使靶基因转录抑制。三个sRNA文库的miRNA这种分布模式,可能暗示着有功能不同或者来源各异的miRNA,参与了对骨骼发育和骨形成关键阶段软骨细胞增殖与分化的调控[1,5]。
但是,总体上看,3个文库中成熟miRNA的种类在的占比却仅仅只有0.69%、0.78%和0.63%,只是整个sRNA种类的极小部分;有一半以上的unique sRNA序列无法与miRBase20.0和Rfam9.1匹配,在基因组上找不到匹配位点,说明这些未知的unann sRNA可能是外源的,或者是通过复杂的转录后加工、修饰而形成的[2]。这些都提示:sRNA的组成、来源以及功能是极其复杂的,我们对于sRNA的认识才刚刚开始。本研究为sRNA调控软骨细胞分化的分子机制提供了大量线索和证据。
另外,由于生物信息学预测标准模糊、算法不同产生了大量假阳性数据和pre-miRNA长度的可变性使得预测结果必然存在遗漏,而且生物信息学无法预测没有基因组或转录组数据库信息的物种和物种特异的sRNA,所以需要生物信息学分析与实验结合对结果进行验证[17-18]。
[1]NGREITZ JM,SIROKMAN K,MCDONEL P,et al.RNARNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites[J].Cell,2014,159:188-199.
[2]HWARTZ S,BERNSTEIN DA,MUMBACH MR,et al.Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA[J].Cell,2014,159:148-162.
[3]RANOV P,OZSOLAK F,KIM SW,et al.New class of genetermini-associated human RNAs suggests a novel RNA copying mechanism[J].Nature,2010,466:642-646.
[4]HUTVAGNER G,MCLACHLAN J,PASQUINELLI AE,et al.A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA[J].Science,2001,293:834-838.
[5]LAURESSERGUES D,COUZIGOU JM,CLEMENTE HS,et al.Primary transcripts of microRNAs encode regulatory peptides[J].Nature,2015,520(7545):90-93.
[6]COLLINS LJ.The RNA infrastructure:an introduction to ncRNA networks[J].Adv Exp Med Biol,2011,722:1-19.
[7]COKUS SJ,FENG S,ZHANG X,et al.Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning[J].Nature,2008,452:215-219.
[8]SUN J,ZHONG N,LI Q,et al.MicroRNAs of rat articular cartilage at different developmental stages identified by Solexa sequencing[J].Osteoarthr Cartilage,2011,19:1237-1245.
[9]ZHONG N,SUN J,MIN Z,et al.MicroRNA-337is associated with chondrogenesis through regulating TGFBR2 expression[J].Osteoarthr Cartilage,2012,20:593-602.
[10]LI R,LI Y,KRISTIANSEN K,et al.SOAP:short oligonucleotide alignment program[J].Bioinformatics,2008,24:713-714.
[11]GIBBS RA,WEINSTOCK GM,METZKER ML,et al.Genome sequence of the Brown Norway rat yields insights into mammalian evolution[J].Nature,2004,428:493-521.
[12]HAFNER M,LANDGRAF P,LUDWIG J,et al.Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing[J].Methods,2008,44:3-12.
[13]GLAZOV EA,COTTEE PA,BARRIS WC,et al.A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach[J].Genome Res,2008,18:957-964.
[14]LU C,JEONG DH,KULKARNI K,et al.Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs(nat-miRNAs)[J].PNAS,2008,105:4951-4956.
[15]CHEN X,LI QB,WANG J,et al.Identification and characterization of novel amphioxus microRNAs by Solexa sequencing[J].Genome Biol,2009,10:R78.2-R78.13.
[16]FOX JG,ANDERSON LC,LOEW FM.Laboratory Animal Medicine[M].San Diego:Academic Press,2002:1265-1325.
[17]CZECH B,MALONE CD,ZHOU R,et al.An endogenous small interfering RNA pathway in Drosophila[J].Nature,2008,453:798-802.
[18]ADAI A,JOHNSON C,MLOTSHWA S,et al.Computational prediction of miRNAs in Arabidopsis thaliana[J].Genome Res,2005,15:78-91.
