CHF2CF2CH2OCHF2与OH自由基反应机理的理论研究
2014-11-27张婷,王丽
张 婷,王 丽
(河南大学 化学化工学院,环境和分析科学研究所,河南 开封 475004)
氟利昂(CFCs)能破坏臭氧层并引起温室效应[1-2],因此寻找氟利昂的替代品迫在眉睫.部分氟氯取代的烷烃化合物(HCFCs)作为氟利昂的一种替代品已被广泛的应用于工业生产的各个领域,但是大部分的HCFCs中仍然含有Cl原子,依然会破坏臭氧层.出于性能、安全性和环境友好等综合因素的考虑,寻找更合适的氟利昂替代品仍然是新世纪科学家所面临的一个巨大挑战.氢氟醚(HFEs)不含有Cl和Br原子因此不会破坏臭氧层,而成为CFCs新一类的替代品被应用于灭火剂、发泡剂、制冷剂和清洁剂等.氢氟醚类化合物除了含有C-H键以外,还含有-O-键,这将提高他们在对流层中的反应活性[3],减少进入平流层的几率,因此其大气寿命较短.在对流层中,HFEs主要与OH自由基发生反应,所以其大气寿命主要由他们与OH自由基反应的速率常数所决定[4].估算HFEs的大气寿命对评估其作为CFCs替代品的可行性,了解其对全球变暖以及对环境的影响是非常重要的.
TOKUHASHI等人[5]测定了CHF2CF2CH2OCHF2与OH自由基反应在250~430K温度区间的速率常数,并给出了该温度区间的阿累尼乌斯公式:kT(250~430K)=2.49×10-12exp[-(1 500±170)/T](单位:cm3·molecule-1·s-1).然而,他们只得到了反应的总速率常数,没有得到详细的反应机理,并且从实验上确定氢原子来自哪个位置是相当困难的,因此,准确的理论计算无疑是解决这个问题行之有效的方法之一.CHF2CF2CH2OCHF2与OH自由基的反应存在两种反应类型,提氢反应和取代反应.可发生取代反应的位置有2个,而可提取的氢原子多达4个,具体的反应通道为:

由于考虑CHF2CF2CH2OCHF2分子具有CS对称性,H2和H3(见图1)是相同的,所以只存在三个氢提取反应通道.为了阐明该反应的反应机理,我们在BMK/6-311+G(d,p)水平下优化了反应的几何结构,并在BMC-CCSD水平下基于优化好的几何结构计算了反应的能量.
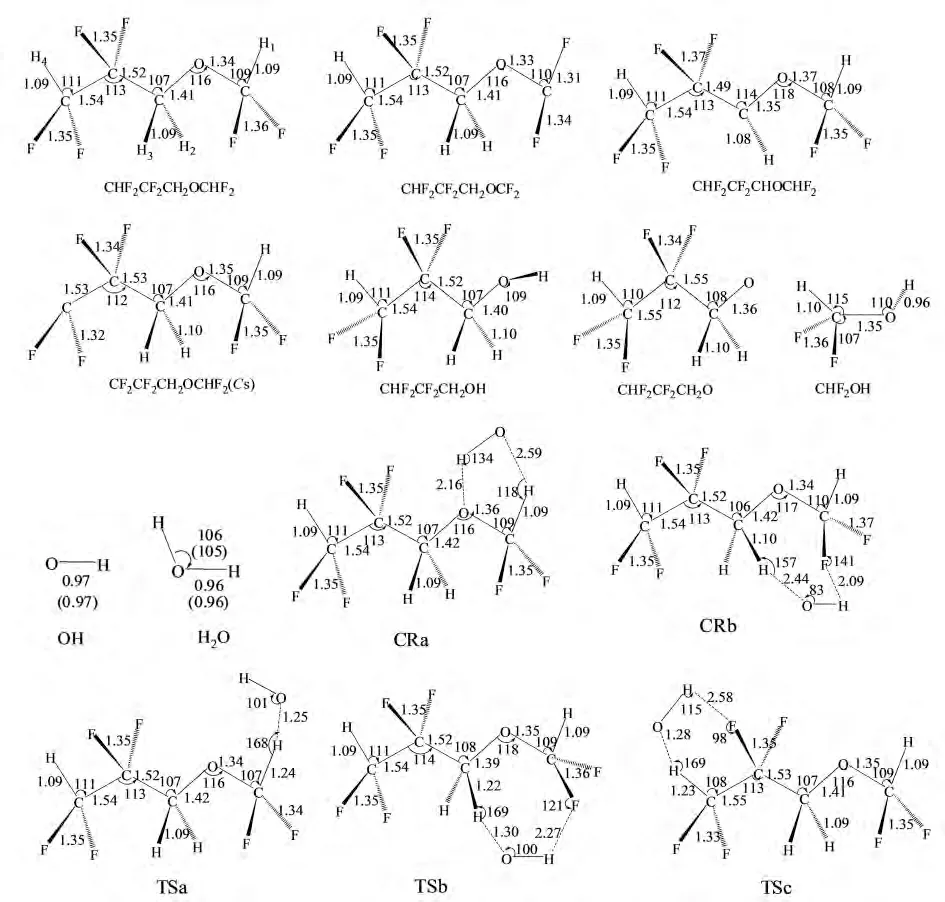
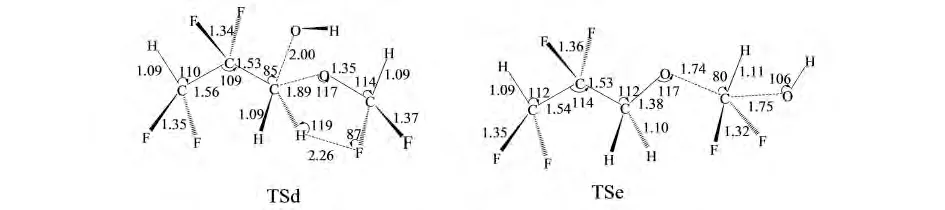
图1 在BMK/6-311+G(d,p)水平下优化的反应物、产物、络合物和过渡态的几何构型Fig.1 Selected optimized geometries of reactant,product,complexes and transition states at the BMK/6-311+G(d,p)level
1 计算方法
首先,利用BMK[6]方法以6-311+G(d,p)为基组优化了反应物、络合物、过渡态以及产物的几何构型并且计算了其振动频率.计算驻点的振动频率有两个作用,一是根据虚频的个数确定优化的几何构型是稳定点还是过渡态,虚频个数为0是稳定点,虚频个数为1是过渡态;二是获得驻点的零点能校正值(ZPE);其次,在同样的水平下,利用内禀反应坐标理论(IRC)[7]获得了反应的最小能量途径(MEP),以确定所找到的过渡态确实连接的是反应物和产物;最后,为了获得更为可靠的反应焓和反应能垒,基于上述优化得到的构型,在BMC-CCSD[8]水平下计算了单点能.
2 结果和讨论
2.1 稳定点的性质
图1绘出了BMK/6-311+G(d,p)水平下优化的所有稳定点(反应物、络合物、产物和过渡态)的几何构型参数以及OH和H2O的实验值[9].在此水平下计算的OH和H2O的几何构型和实验值符合的很好,最大误差为0.21%.在三个氢提取反应的过渡态TSa,TSb和TSc中,即将形成的O-H键相对于孤立H2O分子中相应的键长分别伸长了30.48%,36.01%和33.19%;而即将断裂的C-H键相对于分子CHF2CF2CH2OCHF2中相应的键长分别伸长了13.62%,11.88%和13.12%.可以看出,即将形成的键的伸长程度大于即将断裂的键的伸长程度,表明这三个过渡态构型与反应物构型类似,都是“早垒型”过渡态.而两个取代反应通道的过渡态TSd和TSe都接近于“对称型”过渡态.根据HAMMOND假设[10],我们推断,提氢反应通道应该具有较低的反应能垒而且为放热反应,而取代反应通道应该具有较高的能垒且为吸热反应.
在BMK/6-311+G(d,p)水平下计算得到的反应物、产物、络合物以及过渡态的最小五个振动频率以及相应的实验值[11-12]列在表1中.可以看出,OH和H2O振动频率的计算值与实验值符合得很好,最大误差在7.3%以内.反应物,产物和络合物的频率全部为实频;而过渡态仅有一个虚频.
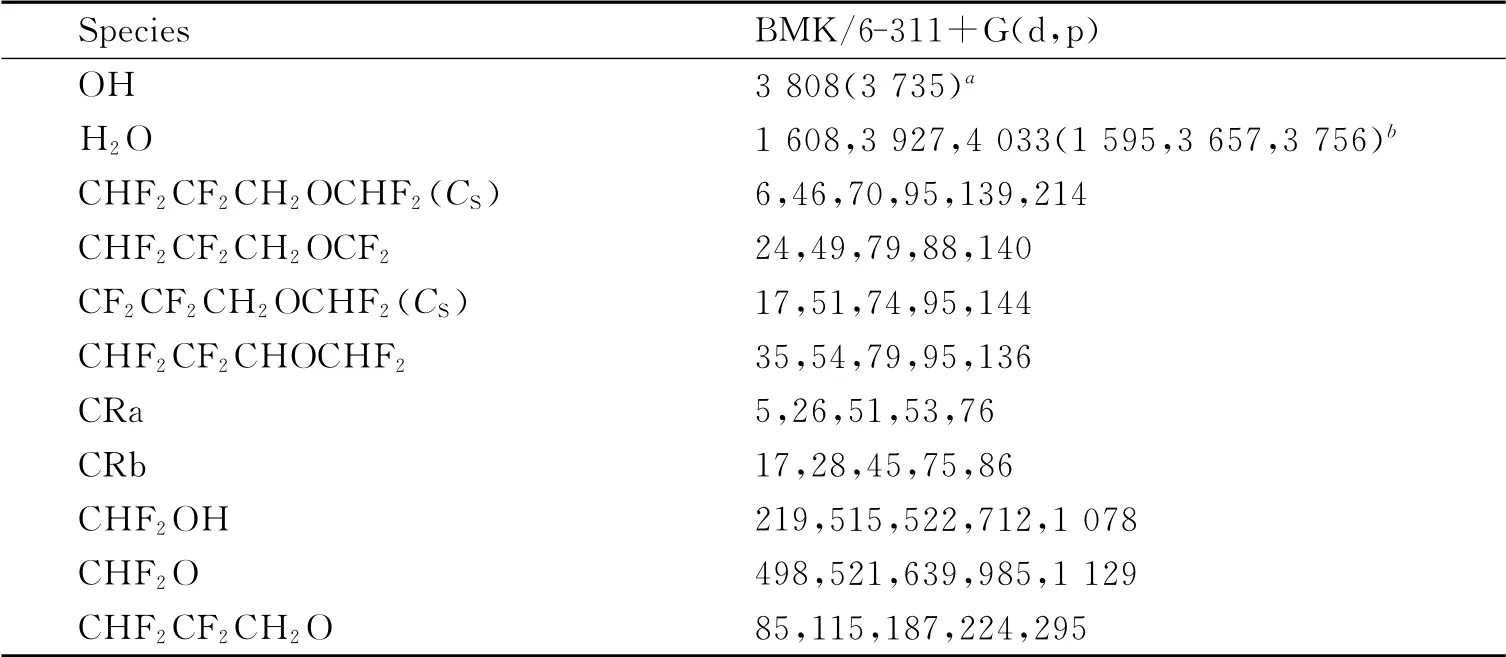
表1 在BMK/6-311+G(d,p)水平下计算的反应物、产物、络合物和过渡态的振动频率(单位:cm-1)Table 1 Frequencies(in cm-1)of the reactants,products,complexes,and transition states calculated at the BMK/6-311+G(d,p)level

续表1
2.2 反应机理和能量
表2列出了BMK/6-311+G(d,p)和BMC-CCSD//BMK/6-311+G(d,p)水平下的反应焓
表2 不同水平下计算的反应焓(ΔH0298)以及相应的实验值(单位:kcal·mol-1)Table 2 Reaction enthalpies at 298K(in kcal·mol-1)calculated at different levels along with available experimental values

表2 不同水平下计算的反应焓(ΔH0298)以及相应的实验值(单位:kcal·mol-1)Table 2 Reaction enthalpies at 298K(in kcal·mol-1)calculated at different levels along with available experimental values
aFrom Refs.[13-15]
Levels BMK/6-311+G(d,p)BMC-CCSD//BMK/6-311+G(d,p) Expt.a Ra -11.70 -14.93 Rb -17.30 -20.24 Rc -12.52 -15.84 Rd -2.55 0.11 Re -4.43 -1.13 CF3CH3+OH→CF3CH2+H2O -9.87 -14.04 -12.11±4 CH3OCH3+OH→CH3OCH2+H2O -20.99 -24.32 -23.11±1.12
由于反应中涉及的绝大多数分子都缺乏相应的标准摩尔生成焓,因此无法将计算值和实验值直接进行比较.我们在相同水平下计算了两个相似反应(CF3CH3+OH→CF3CH2+H2O和CH3OCH3+OH→CH3OCH2+H2O)的反应焓,这两个反应的反应焓有相应的实验值,以此来估算理论计算的准确性.表2中的实验值是通过一系列标准生成焓数据推导出来的,这些标准生成焓包括:OH 9.3kcal·mol-1,H2O-57.81kcal·mol-1,CF3CH3(-179±2)kcal·mol-1,CF3CH2(-124±2)kcal·mol-1,CH3OCH3(-44.00±0.12)kcal·mol-1,CH3OCH2(0±1)kcal·mol-1[13-15].可以看出,对于两个类似的反应,高水平下计算的反应焓和实验值符合的很好,因此有理由推断在相同方法下计算的标题反应的反应焓是可信的.从表2中还可以看出,在高水平下所有的提氢反应通道都是放热反应,而两个取代反应通道是吸热反应或者接近是吸热反应,这与HAMMOND假设[10]的推测是一致的.另外,从热力学上来说,提氢反应比取代反应更容易进行.根据EVANS和POLANYI的假设[16],原子迁移反应的能垒随着放出热量的增加而减少,因此我们可以推测反应Rb应该具有较小的反应能垒,而取代反应相对于提氢反应则具有较高的反应能垒.
图2绘出了反应经零点能(ZPE)校正的势能面示意图.反应物的能量被定义为零点.
对于反应通道Ra,当OH自由基进攻-OCHF2基团上的氢原子时,在氧原子和氢原子之间形成了氢键,因而其存在一个前期络合物CRa.接着,反应经历过渡态TSa后到达产物CHF2CF2CH2OCF2+H2O.Rb通道具有相似的反应机理.而反应通道Rc不存在任何络合物,反应物直接经历一个过渡态TSc到达产物.从势能面上可以看出,两个取代反应的能垒(62.51和50.74kcal·mol-1)远远高于提氢反应的能垒.因此从动力学的观点看,提氢反应通道将是主要反应通道.
由上可知,无论从热力学还是从动力学的观点考虑,提氢反应通道都优于取代反应通道,因此取代反应在总反应中扮演的角色并不重要.对于三个提氢反应通道来说,反应Rb的能垒是三个提氢反应通道中最小的,分别比反应Ra和Rc的能垒小1.11和1.94kcal·mol-1,这符合EVANS和POLANYI假设.综上所述,Rb反应通道,也即提取亚甲基上的氢原子为主要的反应通道.如果用公式Ea,298=ΔE*+nRT=V*+ΔZPE+ΔE(T)+nRT来估算反应的活化能[17],(其中V*,ΔZPE,R,T,ΔE(T)和n分别代表计算势垒、零点能校正、气体常数、热力学温度、热能校正和物质的量),在BMC-CCSD水平下,三个提氢反应通道的活化能分别为1.64,0.03和1.16kcal·mol-1,与TOKUHASHI等人[5]测得的实验值(0.358±0.04)kcal·mol-1之间的误差在合理的范围之内,因此可以相信在BMC-CCSD//BMK/6-311+G(d,p)水平下得到的反应机理是可靠的.

图2 在BMC-CCSD//BMK/6-311+G(d,p)+ZPE水平下反应的势能面示意图Fig.2 Schematic potential energy surface at theBMC-CCSD//BMK/6-311+G(d,p)+ZPE level
3 结论
本文作者运用从头算和密度泛函方法研究了CHF2CF2CH2OCHF2与OH自由基反应的反应机理.计算结果表明,无论从动力学还是从热力学观点看,提氢反应通道都是主要的反应通道,取代反应由于具有较高的能垒而对总反应的贡献可以忽略不计.在三个提氢反应通道中,Rb通道,也即从-CH2-基团上提取氢原子的反应通道是主要的反应通道.
[1]LANGBEIN T,SONNTAG H,TRAPP D,et al.Volatile anaesthetics and the atmosphere:atmospheric lifetimes and atmospheric effects of halothane,enflurane,isoflurane,desflurane and sevoflurane[J].Brit J Anaesth,1999,82(1):66-73.
[2]BURM A G L.Environmental safety in anaesthesia:past and future[J].Curr Anaesth Crit Care,2000,11(3):159-165.
[3]WALLINGTON T J,SCHNEIDER W F,SEHESTED J,et al.Atmospheric chemistry of HFE-7100(C4F9OCH3):reaction with OH radicals,UV spectra and kinetic data for C4F9OCH2·and C4F9OCH2O2radicals,and the atmospheric fate of C4F9OCH2O radicals[J].J Phys Chem A,1997,101(44):8264-8274.
[4]MELLOUKI A,TETON S,LE BRAS G,Kinetics of OH radical reactions with a series of ethers[J].Int J Chem Kinet,1995,27(8):791-805.
[5]TOKUHASHI K,TAKAHASHI A,KAISE M,et al.Rate constants for the reactions of OH radicals with CH3OCF2CHF2,CHF2OCH2CF2CHF2,CHF2OCH2CF2CF3and CF3CH2OCF2CHF2over the temperature range 250-430K[J].J Phys Chem A,2000,104(6):1165-1170.
[6]BOESE A D,MARTIN J M L.Development of density functionals for thermochemical kinetics[J].J Chem Phys,2004,121(8):3405-3416.
[7]FUKUI K.The path of chemical reactions-the IRC approach[J].Acc Chem Res,1981,14(12):363-368.
[8]LYNCH B J,ZHAO Y,TRUHLAR D G.The 6-31B(d)basis set and the BMC-QCISD and BMC-CCSD multicoefficient correlation methods[J].J Phys Chem A,2005,109(8):1643-1649.
[9]LIDE D R.CRC handbook of chemistry and physics[M].80th ed.New York:CRC Press,1999.
[10]HAMMOND G S.A correlation of reaction rates[J].J Am Chem Soc,1955,77(2):334-338.
[11]CHASE M W.NIST-JANAF themochemical tables[M].4th ed.Washington,D.C.and Woodbury,NY:ACS Press,1998.
[12]SHIMANOUCHI T.Tables of molecular vibrational frequencies consolidated volume I[J].J Phys Chem Ref Data,1972,2(1):121-161.
[13]DEMORE W B,SANDER S P,GOLDEN S P,et al.Chemical kinetics and photochemical data for use in stratospheric modeling[M].California:JPL Publication,1997:4.
[14]PILCHER G,PELL A S,COLEMAN D J.Measurements of heats of combustion by flame calorimetry.Part 2-dimethyl ether,methyl ethyl ether,methyl n-propyl ether,methyl isopropyl ether[J].Trans Faraday Soc,1964,60:499-505.
[15]LUO Y R.Comprehensive handbook of chemical bond energies[M].Boca Raton:CRC Press,2000.
[16]EVANS M G,POLANYI M.Inertia and driving force of chemical reactions[J].Trans Faraday Soc,1938,34:11-24.
[17]SCHAFFER F,VEREVKIN S P,RIEGER H J,et al.Geminal substituent effects,15.Enthalpies of formation of a series of fluorinated hydrocarbons and strain-free group increments to assess polar and anomeric stabilization and strain[J].Liebigs Ann,1997,7:1333-1344.
