膜萃取-气相色谱/微分离子迁移谱检测水中的1,4-二恶烷
2014-08-03梁茜茜王卫国李海洋
梁茜茜, 陈 创, 王卫国, 李海洋*
(1.中国科学院大连化学物理研究所,辽宁大连116023;2.大连大学,辽宁大连116622)
1,4-二恶烷(1,4-dioxane),简称为二恶烷,常用作氯代烃溶剂或者其他挥发性有机化合物的稳定剂[1]。氯代烃类污染物是地下水污染中最常见的污染物之一。由于二恶烷在水或有机溶剂中的高溶解性,因此比三氯乙烯(trichloroethylene,TCE)和四氯乙烯(perchloroethylene,PCE)等氯代烃污染物更快且更深地转移入地下水中,造成更严重的水污染。这些污染物直接危害人类的肝脏、肾脏、心脏甚至影响胚胎发育造成婴儿的畸形[2]。二恶烷有潜在致癌性,国际癌症研究机构(IARC)将它划分为2B类[3],美国环保署(EPA)规定饮用水中的二恶烷质量浓度需控制在3 μg/L以下。因此,实现水中二恶烷和氯代烃污染物的在线、灵敏、准确的检测对确保人们生活用水的安全具有重大意义。
检测二恶烷的传统方法为先通过特定样品前处理技术提取二恶烷,再由气相色谱(GC)分离,然后由质谱/氢火焰离子化检测器检测[4,5]。目前,对二恶烷的样品前处理常见的方法有液液萃取[4]、吹扫捕集[4,6]、直接液体注射进样[4]、固相萃取[7]/固相微萃取[8]、顶空[9]/顶空-固相微萃取[7,10]等方法。这些常规方法有着复杂的样品预处理方法与流程,包括野外取样、运输以及实验室仪器分析。近年来,膜萃取(membrane extraction,ME)作为在线检测的进样技术逐渐被人们关注。它利用膜技术将待测物从水相萃取到气相中并由载气输入仪器中完成一步自动化测定,省去样品的采样和运输,节省时间[11]。
微分离子迁移谱(differential mobility spectrometry,DMS)作为一种工作于大气压下的快速离子分离识别技术,发展已有二十余年。自问世以来,以其分析速度快、体积小、功耗低等特点而被广泛 应 用[12,13]。 DMS 基 于 在 高 (Emax≥ 20 000 V/cm)、低(Emin≤1 000 V/cm)变电场作用下离子的迁移率常数变化率的不同对离子进行筛分,使得离子在一个周期内(该周期内射频(radio-frequency,RF)电压对时间的积分为零)运动产生净位移[12]。这时,如果不施加一个补偿电压(compensation voltage,CV)对其进行位移修正补偿,则离子将因偏移位置不断累积而撞击到电极板上被中和掉,不能被检测。DMS可以在高湿度情况下很好地工作,而且有较短的响应时间(0.1 ms),对检测水中的二恶烷有很大的潜力。色谱是最常见的分离复杂样品中化合物的化学分析仪器[14]。利用 GCDMS仪器的二维分离手段,即GC的保留时间和DMS的补偿电压,可以增强对目标化合物的识别准确性。Jochmann等[15]用GC-DMS对40种挥发性有机化合物(volatile organic compounds,VOCs)(包括PCE、苯、氯仿等)进行了检测研究。
在本文中,我们介绍一种膜萃取-气相色谱/微分离子迁移谱装置(ME-GC/DMS)用于检测水中痕量的1,4-二恶烷和氯代烃,并考察了射频电压、补偿电压、采样流速、膜渗透时间、Trap预富集时间等参数对二恶烷检测的影响规律。
1 实验部分
1.1 膜萃取-气相色谱/微分离子迁移谱装置
图1展示了膜萃取-气相色谱/微分离子迁移谱示意图,主要包括膜萃取装置、预富集装置和GC/DMS分析器。膜萃取装置的作用是将二恶烷和卤代烃等污染物从水相中转化成蒸气萃取入膜内,并随着50~300 mL/min管状膜装置的载气送入到预富集装置中。膜装置是由聚二甲基硅氧烷(PDMS)膜(VWR®,Co.)螺旋固定在250 mL石英玻璃瓶内搭建而成[11]。其中膜的长度为50 cm,内径为1.47 mm,壁厚0.23 mm。预富集柱中装填有炭黑吸附剂。气体传输管线的温度保持在100℃;富集温度设置为30℃、富集时间为50~300 s,解吸温度为250℃。富集柱中吸附的分析物,在预富集结束后由载气携带入GC/DMS进行分离和检测。

图1 ME-GC/DMS的装置流程图Fig.1 Schematic diagram of ME-GC/DMS instrumentation setup
GC/DMS分析器为商品化仪器(Sionex,Co.)。其中,气相色谱分离使用VF-624色谱柱(内径250 μm,长度 5 m,Varian公司(Palo Alto)生产)。色谱柱的初始温度设置为 50℃,以 25℃/min的升温速度升至100℃。色谱柱中的馏分依次进入DMS中二次分离和检测。DMS包括一个5 mCi的63Ni放射性离子源、两个平板电极以及用于接受正负离子的两个离子接收电极板。一个平板电极叠加射频电压(RF)和一个直流补偿电压(CV),另一个平板电极接地。在DMS工作的过程中,射频电压的扫描频率为1.25 MHz;补偿电压从+15 V扫描到-45 V,扫描步长为0.55 V,扫描频率1 Hz。DMS的温度保持在120℃。在GC/DMS分析器中,样品按照GC保留时间和DMS补偿电压两个维度进行分离。
1.2 标准样品的配制
实验中使用的标准样品包括1,4-二恶烷、二氯乙烯(dichloroethylene,DCE)、TCE、PCE 及四氯化碳等均购自Sigma Aldrich(Milwaukee,WI),均为色谱纯(>99.9%)。每一种样品用去离子水配制质量浓度为1 000 μg/L的母液,实验过程中所需不同浓度的样品溶液均为逐级稀释这些母液得到。
2 结果与讨论
2.1 实验条件对二恶烷检测的影响
2.1.1 射频与补偿电压
图2展示了DMS对100 μg/L二恶烷检测时的分辨能力和响应强度随着射频电压幅值变化的趋势图。分辨能力(Rd)的计算公式为:Rd=CV/Wh/2,式中,CV为对目标离子检测的补偿电压,Wh/2则为在该补偿电压下的半峰宽。从图2中我们可以观察到,当RF的幅值电压由600 V增加到1 100 V时,二恶烷的分辨能力随之由0.67增加到3.13。然而,二恶烷的响应强度却随着RF升高而降低,即从0.71 a.u.下降到了0.07 a.u.。检测灵敏度降低的原因可能是在高射频电压下,离子在一个射频周期内在纵轴上产生的净位移程度增加,更易撞击到电极板上被中和掉,从而造成更严重的离子信号损失。综合考虑检测的分辨率与灵敏度的影响,1 000 V被选为优化的射频电压幅值。
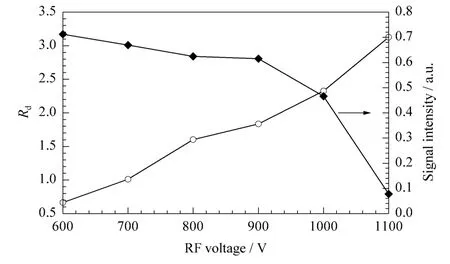
图2 二恶烷的分辨率与响应强度随着射频电压幅值变化的趋势图Fig.2 Plots of resolution and signal intensity of dioxane versus maximum RF voltage,respectively
图3展示了对二恶烷检测时,所需的补偿电压随射频电压(600~1 000 V)变化的趋势图。显而易见,补偿电压的绝对数值随着射频电压的增加而增大。因为随着射频电压的增大,离子在一个周期内运动产生的纵轴净位移也会增加,因此要使离子顺利通过电极板,必须要随之增大补偿电压。在选定的1 000 V射频电压下,其对应的补偿电压-3.3 V即为二恶烷的特征值。

图3 检测二恶烷的补偿电压随着射频电压变化的趋势图Fig.3 Plot of compensation voltage versus the maximum RF voltage for dioxane
2.1.2 采样流速与膜渗透时间
图4给出了100 μg/L二恶烷的PDMS膜连续萃取结果。可以观察到,二恶烷的渗透通量随着时间延长先是迅速的上升,而后逐渐趋于饱和。这可能是由于样品二恶烷的浓度随着萃取的进行不断地降低,膜两侧二恶烷的浓度差在时刻发生着变化,因此膜相内部也并没有达到渗透平衡时的浓度差分布造成的。图4还显示了 GC/DMS分别在50 mL/min和150 mL/min载气流速的吹扫下,对100 μg/L二恶烷的响应强度达到最大值所需要的时间均超过30 min,且载气流速越小渗透达到平衡的时间越长。
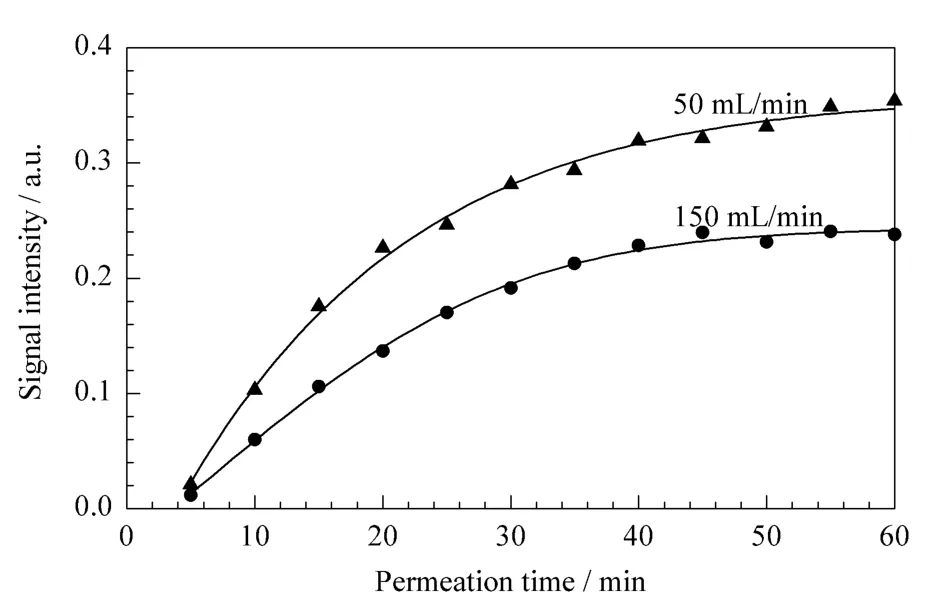
图4 不同采样气流速下GC/DMS对100 μg/L二恶烷的响应强度随膜渗透时间变化的趋势图Fig.4 Plot of signal intensity of GC/DMS versus permeation time for 100 μg/L 1,4-dioxane at different gas flow rates
图5给出了100 μg/L二恶烷在膜中的响应强度随载气流速变化的情况。实验发现,在膜萃取达平衡后,随着膜内载气流速由50 mL/min增加至300 mL/min,反而导致了二恶烷响应强度的降低。这说明随着流速的增加,蒸发过程加强,载气的稀释作用开始体现出来,从而使二恶烷的浓度下降。综合考虑膜内载气的流速与萃取时间的影响,我们选择50 mL/min膜内载气流速及渗透30 min作为ME-GC/DMS富集柱开始采样的时间。
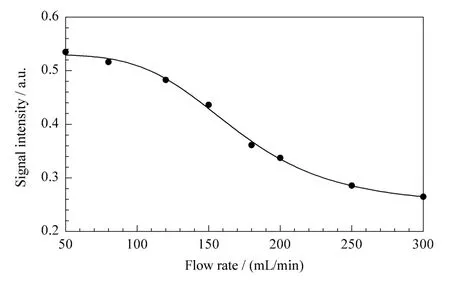
图5 采样载气流速对100 μg/L二恶烷响应强度的影响Fig.5 Effect of gas flow rate on signal intensity for 100 μg/L dioxane
2.1.3 预富集时间
图6展示了ME-GC/DMS系统在不同富集时间下100 μg/L二恶烷的信号响应强度变化的趋势图。可以看到随着富集时间的延长,二恶烷的响应强度呈现上升的趋势。考虑到富集柱中固定量的吸附剂会有最大吸附量,我们推测这种上升的趋势会随着富集时间的进一步延长而呈现出饱和的状态。与预期一样,ME-GC/DMS对二恶烷的响应强度随着富集时间的增长,在150 s以后趋于饱和。因此,我们将150 s的预富集时间用于后续的实验中。
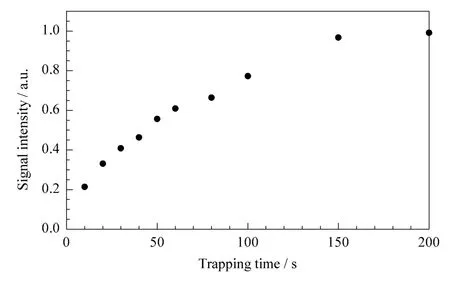
图6 GC预富集装置的富集时间对二恶烷响应强度的影响Fig.6 Effect of trapping time on signal intensity of dioxane
2.2 二恶烷的动态响应
在优化的条件下,即射频电压为1 000 V,补偿电压为-3.3 V,采样流速为50 mL/min,膜渗透时间为30 min,预富集时间为150 s,定量检测二恶烷。图7展示了二恶烷的正离子峰高与质量浓度之间的关系曲线。其插图表明,二恶烷在2~20 μg/L范围内呈良好的线性关系,相关系数(r)为0.992。通过计算(S/N=3)得到二恶烷的检出限(LOD)为0.67 μg/L。通过将100 μg/L 和10 μg/L 二恶烷添加在自来水中进行6次平行测定,获得的平均回收率分别为105.03%和87.05%;RSD分别为4.9%和3.6%。表明该方法具有较好的准确度与重现性,有望应用于地下水中二恶烷的实时监测。
与文献报道的二恶烷分析方法相比较,本文采用的ME-GC/DMS方法的LOD与线性范围都与现有技术相当。例如:Li等[16]利用冷冻微萃取进样技术结合GC/MS获得二恶烷的检出限为1.6 μg/L;Nakamura等[17]利用顶空固相微萃取GC/DMS测定二恶烷的线性范围为5~100 μg/L,检出限为1.17 μg/L。此外,我们建立的方法具有自身的优势,即我们的膜萃取装置可以实现一步检测,从而可对水样中的二恶烷进行在线分析。

图7 二恶烷(2~500 μg/L)的浓度-响应曲线图Fig.7 Ion intensity versus the mass concentration of dioxane(2 to 500 μg/L)in water
2.3 二恶烷与氯代烃混合物的检测
图8展示了使用 ME-GC/DMS检测50 μg/L 1,4-二恶烷与5种氯代烃的混合样品所得到的二维等高线图。DMS能够同时输出正负离子信号,因此图8包括了正离子通道和负离子通道的二维谱图。在图8中,纵轴表示分析物在色谱中的保留时间,横轴是DMS的补偿电压,颜色代表峰强度。表1列出了二恶烷和5种氯代烃在ME-GC/DMS中的正负离子信息。
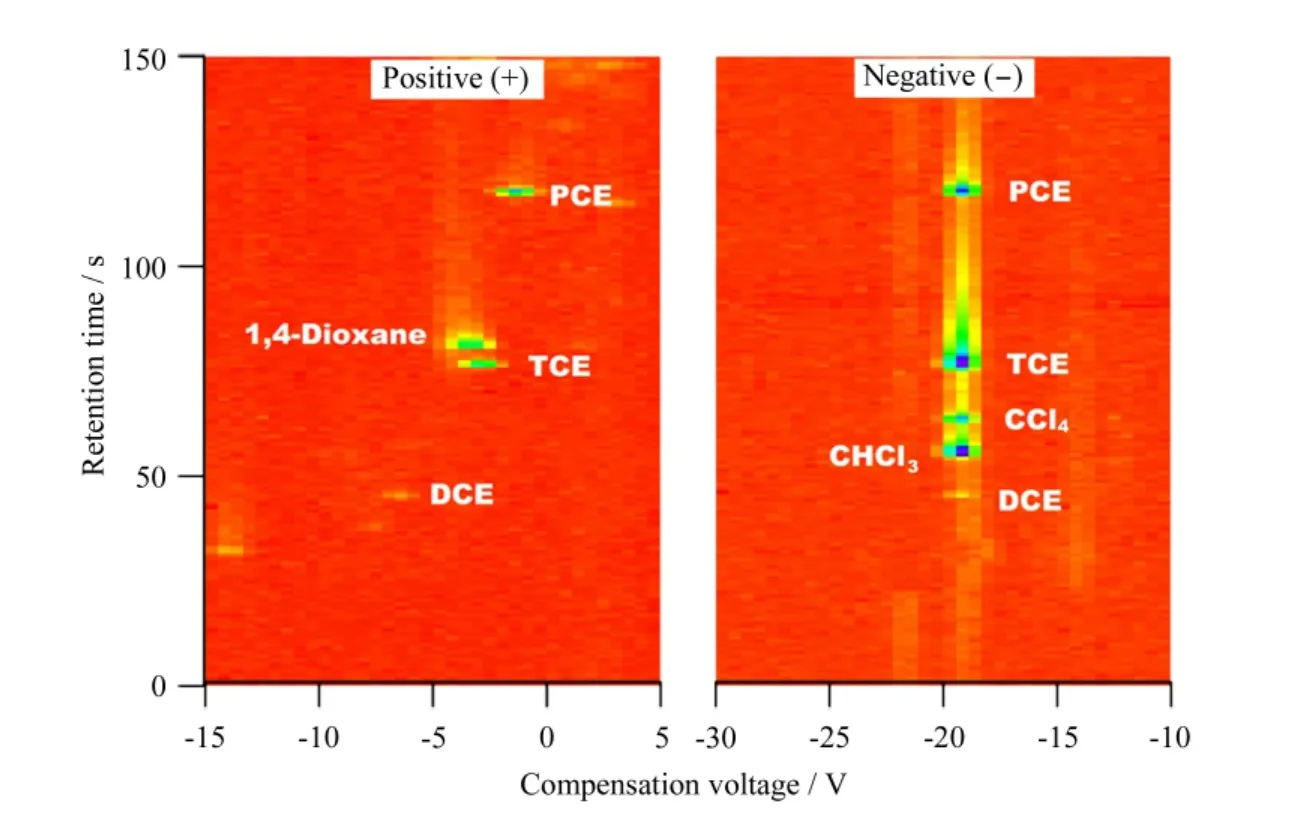
图8 GC/DMS对水中的二恶烷和5种氯代烃混合物的正、负离子通道二维分离图Fig.8 Two-dimensional plots from ME-GC/DMS analysis of 1,4-dioxane with five aqueous chloride mixture in both the positive and the negative ion channels
首先,在正离子通道中我们观察到4个离子峰。按照色谱法保留时间的先后依次为DCE、TCE、二恶烷、PCE。其中,我们的目标分析物二恶烷的二维坐标(CV,tR)为(-3.33 V,95.04 s)。其他氯代烃混合物不会对二恶烷的识别造成干扰。1,4-二恶烷具有较高的质子亲和势(PA=797.4 kJ/mol)[1],与试剂离子H3O+(H2O)n发生质子转移生成正产物离子。而DCE、TCE、PCE由于具有较低的质子亲和势[18,19],不能通过质子转移生成正产物离子,我们推测DCE、TCE、PCE的正离子产物的形成可能是通过与大气压化学电离源中的初始离子发生电荷转移反应生成的[20,21]。

表1 GC/DMS对1,4-二恶烷及氯代烃检测的二维参数Table 1 Results for different chemicals with GC/DMS
在负离子通道中离子峰呈现出垂直簇状分布特征,可以观察到色谱保留时间不同但是具有相同补偿电压的氯代烃化合物的峰。这些氯代烃化合物的中心补偿电压均为-19.44 V,说明形成了相同的负产物离子Cl-(H2O)n。同时,根据色谱的不同保留时间可以将它们很好地区分开。此外,DCE、TCE、PCE在正负离子通道均有响应,从而给这些化合物身份的确认提供了双重识别信息。
3 结论
以气相色谱/微分离子迁移谱技术结合管状膜萃取进样装置用于水中1,4-二恶烷污染物的检测。通过调节射频电压、优化载气流速、GC预富集时间等参数,建立了检测水中二恶烷污染物的方法和流程,建立了浓度校正曲线。实现了GC/DMS对水中痕量1,4-二恶烷和5种氯代烃的分离和分析。
致谢 本工作在美国橡树林国家实验室完成,感谢徐俊博士的帮助与指导。
[1] NIST Standard Reference Database Number 69.[2014-04-23].http://webbook.nist.gov/chemistry/
[2] Wells G,Schroth W,Brauch H,et al.Clin Nephrol,2009,71(6):708
[3] Grimmett P E,Munch J W.J Chromatogr Sci,2009,47(1):31
[4] Draper W M,Dhoot J S,Remoy J W,et al.Analyst,2000,125(8):1403
[5] Isaacson C,Mohr T K G,Field J A.Environ Sci Technol,2006,40(23):7305
[6] Epstein P S,Mauer T,Wagner M,et al.Anal Chem,1987,59(15):1987
[7] Kawata K,Ibaraki T,Tanabe A,et al.J Chromatogr A,2001,911(1):75
[8] Shirey R E,Linton C M.J Chromatogr Sci,2006,44(7):444
[9] Shin H-S,Lim H-H.Chromatographia,2011,73(11/12):1233
[10] Jochmann M A,Kmiecik M P,Schmidt T C.J Chromatogr A,2006,1115(1):208
[11] Du Y,Zhang W,Whitten W,et al.Anal Chem,2010,82(10):4089
[12] Shvartsburg A A.Differential Ion Mobility Spectrometry:Nonlinear Ion Transport and Fundamentals of FAIMS.Boca Raton,FL:CRC Press,2009
[13] Zhang J,Li L F,Guo D P,et al.Chinese Journal of Analytical Chemistry(张洁,李灵锋,郭大鹏,等.分析化学),2013,41(7):986
[14] Wang J C,Xiong L,Zhang H J,et al.Chinese Journal of Chromatography(王金成,熊力,张海军,等.色谱),2013,31(2):139
[15] Telgheder U,Malinowski M,Jochmann M A.Int J Ion Mobil Spec,2009,12(4):123
[16] Li M,Conlon P,Fiorenza S,et al.Ground Water Monit Remediat,2011,31:70
[17] Nakamura S,Daishima S.Anal Chim Acta,2005,548:79
[18] Kennedy R A,Mayhew C A,Thomas R,et al.Int J Mass Spectrom,2003,223/224:627
[19] Nicoletti A,Paradisi C,Scorrano G.Rapid Commun Mass Spectrom,2001,15(20):1904
[20] Mayhew C A,Thomas R,Watts P.Int J Mass Spectrom,2003,223/224:91
[21] Dotremont C,Goethaert S,Vandecasteele C.Desalination,1993,91(2):177
