利用分析Unigene在转录组中表达模式的方法拼接盐角草铵转运基因
2014-06-24肖薪龙张选吴晓朦马金彪姚银安
肖薪龙,张选,吴晓朦,马金彪,姚银安
1中国科学院新疆生态与地理研究所干旱区生物地理与生物资源重点实验室,新疆乌鲁木齐830011 2中国科学院大学,北京100049
利用分析Unigene在转录组中表达模式的方法拼接盐角草铵转运基因
肖薪龙1,2,张选1,2,吴晓朦1,2,马金彪1,姚银安1
1中国科学院新疆生态与地理研究所干旱区生物地理与生物资源重点实验室,新疆乌鲁木齐830011 2中国科学院大学,北京100049
RNA-seq技术能够全面快速地获得物种在某一状态下的转录本序列信息,但测序并组装后的大量Unigene往往不包含完整ORF(Open reading frame)。转录组库具有一定的冗余性,存在着属于同一个转录本的Unigene,这些Unigene因为无重叠区不能拼接而存在转录组库中。基于这种情况,为了拼接铵转运蛋白家族Unigene,首先挑选注释为AMT(Ammonium transporter)且ORF不完整的所有Unigene(5条),通过分析Unigene在4个转录组的表达模式,其中2条Unigene(Uni4和Uni5)具有相同的表达模式,推测可能来自同一转录本。然后通过NCBI blastx将这2条Unigene与参考物种的AMT蛋白质比对,确定其在转录本的位置及序列相互间没有交叠(如果两条编码序列相互交叠则不能组成同一个转录本)。结果发现Uni4和Uni5分别位于参考转录本5′端和3′端位置,因此假定它们属于同一个转录本,中间空缺约120 bp未知序列。通过试验验证,分别在Uni4和Uni5上设计单正向引物和单反向引物,PCR扩增得到约800 bp片段,将其测序并与两条Unigene比对,证实Uni4和Uni5属于同一转录本且获得了缺失的未知序列。最终拼接得到1 667 bp序列,包含1 482 bp完整ORF,编码494个氨基酸,通过系统进化分析将其归类为am t1亚家族,命名为Seamt1。生物信息学手段预测SeAMT1蛋白与已知的其他物种AMT性质相似。本研究采用转录组Unigene表达模式聚类的方法挖掘潜在的同一转录本Unigene,并且通过另外两组Unigene检验了该方法的可行性。这一便捷方法有助于转录组中Unigene的延伸和拼接,有助于完整ORF的获得及后期基因功能研究。
转录组测序,基因表达,序列组装,克隆方法,RPKM,氮吸收
盐角草Salicornia europaea是一年生双子叶草本植物,茎肉质化,生长于沿海滩涂或内陆潮湿的盐碱地,是一种最耐盐的真盐生植物之一[1]。盐角草不仅具有高抗盐能力和盐富集能力[2],而且具有高效氮肥吸收和利用能力[3]。Webb等利用盐角草作为污水生物滤池,能清除输入污水中(98.2±2.2)%无机氮(NH4+和NO3–),其中NH4+的清除能达到91%以上[4]。盐角草耐盐基因的挖掘得到了人们的重视[5-6],但氮转运基因的研究却很少。
铵态氮(NH4+)是植物氮源之一,通过植物细胞膜上的AMT(Ammonium transporter)转运蛋白进入细胞,最终同化为氨基酸,进入植物体内氮循环[7-8]。AMT是铵转运蛋白的编码基因,在模式植物拟南芥中发现了6个[7],在水稻中至少存在12个[9],而盐角草Seamt基因还未见报道。我们之前的工作将盐角草在不同盐处理及不同组织进行转录组测序[10],为SeAMT基因克隆打下了基础。
转录组指某个物种或特定细胞在某一发育阶段和功能状态下产生的所有RNA的总和,包括mRNA和非编码RNA(Non-coding RNA, ncRNA)[11]。转录组测序(RNA-Seq)是近年来发展起来的一种测序技术,通过新一代高通量测序,能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息[12-13]。转录组测序读段(Read)长度一般较短,Trinity方法的出现使得即使无基因组参考物种的转录组read也可以有效组装为Uingene,甚至组装到全长序列[14]。但是在转录组库中仍然存在大量不包含完整ORF的Uingene。得到包含完整编码区的Unigene序列,是基因功能研究的基础性工作[15]。
将转录组的Unigene片段延伸得到完整ORF全长有以下策略:1)将转录组的Unigene与数据库中该物种EST序列组装(电子克隆)。这个方法对于核苷酸序列丰富的物种可能有效,但是对于非模式物种,特别是核酸序列信息较少的物种,电子克隆方法并不适用[16-17]。2)对于基因组已测序的物种,如拟南芥和水稻,可直接将感兴趣的Unigene与参考基因组进行比对,获取该基因的全部信息,进一步分析其可能的转录本序列。3)RACE技术(cDNA末端快速扩增)可有效地延伸Unigene所缺的5ʹ端或3ʹ端序列[18],然而市场上RACE试剂盒价格昂贵,投入成本较高。
在之前AMT基因克隆试验,我们采用传统的RACE方法克隆,扩增缺失的Unigene 5ʹ端或3ʹ端并测序,结果发现这些序列就是转录库中的某些Unigene。如,Unigene11 473 RACE延伸的5ʹ端序列与Unigene59 692和Unigene76 680序列高度一致(比对结果未显示),它们属于同一转录本(表1)。Unigene142 163,Unigene11 551和Unigene71 089经证实也是属于同一转录本(表1)。因此,转录组库中的Unigene具有一定的冗余性,即属于一个转录本的两个或多个Unigene同时存在。这些Unigene因相互间没有重叠区或其他原因无法拼接为一条转录本[19]。此外,我们发现这些属于同一转录本的Unigene的RPKM(Reads per kilo bases per m illion reads)值存在一定规律——在各个转录组间具有相同的表达模式(图1)。如果能利用表达模式相同这一性质,挖掘来自同一转录本的Unigene,将使得序列拼接及全长基因获得更加容易。为证实该设想的可行性,我们对其他AMT Unigene进行了验证。本文以拼接盐角草Seamt基因为例,介绍一种通过Unigene在各个转录组的表达模式分析、Unigene编码蛋白位置分析、PCR验证的方法,从转录组中拼接属于同一转录本的序列。

图1 已知分别属于两个转录本的Unigene表达模式Fig.1 Expression patterns of the Unigenes which were respectively belonged to two transcripts.
1 材料与方法
1.1 材料
盐角草种子采集于新疆阜康县,温室内人工栽培,苗龄一个月后取样,液氮速冻后存放于–80℃冰箱。
1.2 方法
1.2.1 转录组测序
以200 mmol/L NaCl处理的盐角草为实验组,0 mmol/L NaCl处理为对照组,分别取地上和地下部分,分别标记为转录组1、转录组2、转录组3、转录组4。Illum ina HiSeq2 000转录组测序、Unigene组装及注释、表达量计算等工作依托华大基因公司完成。
1.2.2 Unigene的表达模式及序列分析
从盐角草RNA-Seq的组装结果中挑选注释为AMT的Unigene,通过对这些Unigene进行ORF搜索,排除包含完整ORF的Unigene,余下的不完整Unigene用以后续分析。统计这些不完整Unigene在4个转录组库中的RPKM值,绘制表达模式分析图,根据表达量的升、降、不变这三种情况确认表达模式一致的Unigene。提取表达模式一致的Unigene核苷酸序列,与NCBI参考物种的蛋白质序列比对,确认Unigene所处的转录本位置及Unigene间是否有交叠。相互间有交叠的Unigene可排除以减小工作量,留下没有交叠Unigene做进一步验证。根据Unigene所处位置将这两条或多条Unigene整合成一条FASTA序列,中间可能缺失区域以“N”代替。
1.2.3 PCR实验验证
总RNA提取参照Qiagene试剂盒说明书,cDNA第一条链合成参照反转试剂盒(TaKaRa,大连宝生物)。以两条Unigene的整合序列为模板,用Primer 5.0在连接处的上游和下游200 bp处分别设计正向和反向引物,产物横跨两条Unigene。引物合成(华大基因,北京);高保真2×prem ix PCR试剂(康为公司,北京)PCR扩增;琼脂凝胶电泳检测PCR产物。
1.2.4 测序验证及序列组装
PCR产物由北京华大基因公司测序,测序结果与两条Unigene用NCBI blastn比对,然后CAP3(http://doua.prabi.fr/software/cap3)在线组装。
1.2.5 组装序列的生物信息学分析
利用生物信息学软件及在线工具,分析组装序列开放阅读框ORF(http://www.ncbi.nlm. nih.gov/gorf/gorf.htm l),用MEGA 5.0将组装序列与拟南芥、水稻、小麦、番茄的AMT基因进行系统进化分析,将其初步命名。通过以下方法对组装序列功能进行预测:蛋白基本理化性质分析ProtParam:http://www.expasy.org/tools/ protparam.htm l;亲疏水性分析ProtScale:http:// cn.Expasy.org/tools/protscale.htm l;跨膜区预测TMHMM Server:http://www.cbs.dtu.dk/services/ TMHMM/;信号肽预测SignaIP 3.0 Server: http://www.Cbs.Dtu.dk/services/SignalP-3.0;亚细胞定位预测WoLFPSORT:http://psort.Hgc.jp。生物信息预测结果与其他物种已知AMT蛋白特性进行比较,推测其可能的铵转运功能。
2 结果
2.1 转录组中不完整的AMT Unigene
在盐角草转录组中共发现14条注释为AMT(Ammonium transporter)的Unigene,其中5条Unigene的ORF不完整且无法聚类拼接。分别将其编号为Uni1、Uni2、Uni3、Uni4、Uni5,核酸序列长度分别为1 133 bp、918 bp、267 bp、885 bp、671 bp(表2)。将这5条序列分别在NCBI进行blastx比对,比对结果与转录组注释结果一致,推测Uni1、Uni2、Uni3、Uni4、Uni5都属于AMT家族。

图2 五条Unigene在4个转录组的表达模式Fig.2 Expression patterns of five Unigene in 4 transcriptomes.

图3 低表达基因Uni3,Uni4,Uni5的表达量模式Fig.3 Expression patterns of low expressed genes Uni3,Uni4,Uni5.
2.2 在不同转录组中的表达模式分析
RNA-seq对基因表达量的评估是根据该基因读段(Reads)的数量,数量越多,表达量越高。但是读段数会受基因长度和测序深度的影响,Mortazavi等整合这两个因素提出了RPKM作为表示基因表达量的指标[20]。基因在各个转录组间有3种表达变化,即表达上升、表达下降、表达不变。因此基因在4个转录组间的表达模式总共有27种(3n–1,3表示3种表达变化;n为转录组个数)。5个AMT Unigene在4个转录组的表达模式有3种(图2和图3):1)升—降—升:Uni1和Uni2;2)不变—升—降:Uni3;3)不变—不变—升:Uni4和Uni5。其中Uni1和Uni2表达模式相似,Uni4和Uni5的表达模式几乎一致(图3)。因此,我们推测Uni1和Uni2来自同一基因,Uni4和Uni5来自另一个基因,将其分别聚为一组做进一步分析(Uni1,Uni2本文未做分析,仅以拼接Uni4,Uni5为例)。
2.3 分析Uni4和Uni5在转录本的位置
Uni4和Uni5序列长度分别为885 bp、671 bp,将这两条序列通过NCBI blastx分别与参考物种的AMT蛋白进行比对分析。结果如图4所示,AMT参考物种蛋白约500个氨基酸,Uni4与参考蛋白比对结果(图4A)显示Uni4编码5ʹ端1−282位置氨基酸,5ʹ端ORF完整,而3ʹ端缺失。同理,Uni5与参考蛋白比对结果(图4B)说明Uni5编码3ʹ端324−500位置氨基酸,3ʹ端ORF完整,而5ʹ端缺失。将两条序列整合为一条序列(Uni4在5ʹ端,放前面;Uni5在3ʹ端,放后面),然后与参考蛋白blastx比对,比对结果(图4C)显示整合序列具有完整的5ʹ端和3ʹ端,中间缺失了大约40个氨基酸(120 bp)。因此,我们推测Uni4和Uni5具有组成一个转录本的可能性。
这一步工作是为了证实两条Unigene间确实因为存在着空缺而不能组装在一起。如果这两条Unigene的编码蛋白有交叠,而交叠区的核酸序列相似度不高,不可能组成同一个转录本,应当舍弃,降低工作量。如果两条Unigene没有交叠则有可能来自一条转录本,进行下一步PCR实验验证。
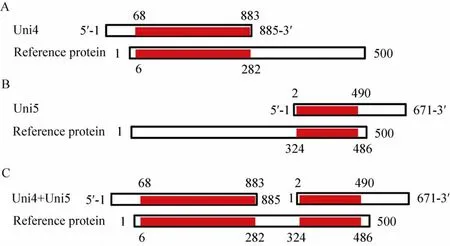
图4 Uni4和Uni5的b lastx比对分析Fig.4 A lignment of Uni4 and Uni5 w ith reference protein by blastx.The red regions indicate matching between Unigene and reference protein.
2.4 PCR扩增验证
分别在Uni4的521−541 bp处设计正向引物(5ʹCTCGCCTACTCCACACTCCTT 3ʹ),在Uni5的341−361 bp处设计反向引物(5ʹGCTCCCCATTGTCACACTCAC 3ʹ)(图5)。以盐角草的cDNA为模板,2×prem ix PCR试剂进行PCR扩增,设两个重复。PCR产物用1.2%琼脂凝胶电泳检测,如图6所示,电泳获得单一条带,大小约800 bp,与预期结果相符。
2.5 测序验证
PCR产物由华大基因(北京)进行双向测序,测序峰图良好。测序序列编号为overlap1,长度809 bp。以overlap1为Query序列与Uni4, Uni5在NCBI进行blastn比对。如图7,overlap1横跨Uni4,Uni5两条序列,相似度分别为99%和100%。Uni4与overlap1在784 bp处存在一个第3位碱基C-G突变,即GTC与GTG,但都编码缬氨酸。测序结果验证了overlap1与Uni4及Uni5同属一个基因。

图5 引物设计示意图Fig.5 Diagram of primer design.FP:forward primer; RP:reverse primer.

图6 PCR产物琼脂糖凝胶电泳Fig.6 Agarose gel electrophoresis of PCR product.M: marker.

图7 over lap1与Uni4,Uni5的blastn比对Fig.7 A lignment beteween overlap1 and Uni4,Uni5 by blastn.
2.6 序列组装及分析
将Uni4,Uni5和overlap1在CAP3[21]网站上在线组装,组装结果编号为Secontig1。对Secontig1进行ORF搜索,发现其包含1 482 bp的ORF序列,编码494个氨基酸,与其他物种的AMT1氨基酸数目相近。Secontig1(登录号KJ487970)序列长度1 667 bp,5ʹ端起始密码子附近符合kozak的A/GNNATGG规则[22],同码框的起始密码子上游具有终止密码子,因此判断该序列具有完整的编码区。用MEGA 5.0软件对Secontig1编码蛋白与模式植物拟南芥和主要作物水稻、小麦、番茄的6个、10个、3个、3个AMT蛋白序列序列进行系统进化分析。23个AMT蛋白可分为两大组:所有AMT1亚家族归为A组;AMT2、AMT3、AMT4亚家族归类为B组。A组可再分为三组,第一组同为禾本科的水稻和小麦AMT1近缘相似度高而归类一起;第二组十字花科拟南芥AMT1.2,AMT1.3-1.5单独归类;第三组番茄LeAMT1.1-1.3,AtAMT1.2,SeAMT1归为一组。B组水稻OsAMT2,OsAMT3及小麦TaAMT2.1可归为一组,AtAMT2和OsAMT4各自单独成一支(图8)。
因此Secontig1归类于am t1亚家族,并与番茄LeAMT1.3相似度最高,将Secontig1命名为Seam t1。

图8 SeAM T1系统进化树Fig.8 Phylogenetic analysis of SeAMT1.At: Arabidopsis thaliana;Os:Oryza sativa;Ta:Triticum aestivum;Le:Lycopersicum esculentum;Se:Salicornia europaea.
2.7 SeAM T1蛋白理化性质分析及功能预测
通过在线网站和工具ProtParam分析SeAMT1蛋白分子量53 224.9,理论等电点5.98,不稳定系数25.88,属于稳定蛋白。ProtScale分析SeAMT1蛋白存在多个亲水区和疏水区,可能与其功能有关;TMHMM Server预测有9个跨膜区,WoLFPSORT预测SeAMT1主要位于质膜,预测分值10.0,而预测位于内质网膜分值只有2.0。SignaIP神经网络模型(NN)预测SeAMT1不具有信号肽,并且马可夫模型(HMM)预测SeAMT1不属于分泌蛋白,支持了WoLFPSORT预测SeAMT1是膜内在蛋白的结论。以上分析预测结果都支持了新方法克隆的Seam t1属于AMT基因家族的成员之一;推测所编码的SeAMT蛋白与其他物种性质相符——亚细胞定位于质膜及NH4+转运功能。
3 讨论
尽管是遗传背景不清楚的生物,RNA-seq测序能提供大量的转录本,为基因克隆提供了极大方便[12]。然而非模式生物(如盐角草等)缺乏基因组信息参考,读段的组装难度增加[23]。一般来说,RNA-seq测序读长越长,越有利于测序片段的装配,而目前Roche 454测序平均读长400 bp,Illum ina平均读长只有100 bp[24]。虽然通过对测序读段的组装可以获得一些全长Unigene,但大部分Unigene不包含完整ORF。转录组Unigene的数量通常在十万级以上,具有一定的冗余性,来自同一转录本的两条或多条Unigene无法组装而同时存在于转录组库中[25]。这些Unigene序列往往不长,只包含转录本的5ʹ端或3ʹ端,并且相互间没有重叠区域。从转录组中发现这些Unigene有助于不完整基因的延伸和拼接。
首先根据转录组注释挑选感兴趣的基因家族中所有非完整Unigene,排除大量非目的基因干扰,然后进行表达模式分析。来自同一个转录本的任意区域序列应具有相同的表达丰度,因此这些Unigene的表达量在理论上是相等的,即表达评估值RPKM是相近的。Unigene在两个以上转录组的表达模式分析可以有效区分来自一个转录本的Unigene,转录组越多,表达模式越丰富,区分效果越好。因此在各个转录组间表达模式一致的Unigene很可能来自同一个转录本。如图1,先前已证实分别属于两个转录本的两组Unigene,它们在转录组间的表达模式具有一致性,支持了本文推论。
当然,即使具有相同的表达模式的Unigene,它们也可能分别来自功能相近或表达相似的不同基因。如图2中的Uni1和Uni2与图1中的Unigene142 163、Unigene11 551和Unigene71 089这3条Unigene有着相同的表达模式,但它们并不是同一个转录本的序列。因此需要做进一步分析来排除干扰。如果几个Unigene的注释信息明显不同,则不太可能是同一个基因;如果Unigene序列间具有一定交叠区域而且无法匹配,说明它们不可能拼接上,因此不必再做PCR验证。总之,先根据基因注释挑选基因,然后将表达模式一致的Unigene聚类分组,排除Unigene间有交叠区域的Unigene组,剩下各个组的两条或多条Unigene最后通过PCR扩增和产物测序验证它们是否来自同一条转录本。
通过生物信息学工具分析,发现所得到的基因Seam t1编码区完整,编码蛋白的氨基酸数目与其他物种一致,是盐角草AMT1亚家族成员之一。TMHMM跨膜区预测认为SeAMT1蛋白有9个跨膜区,而普遍认为植物的AMT蛋白有11−12个跨膜区[26-27],这可能是因为生物信息学预测存在一定偏差或是物种存在的差异。通过SignaIP 3.0在线预测发现SeAMT1蛋白具有信号肽可能性较低,不会是分泌蛋白[28];而WoLFPSORT亚细胞定位预测它位于质膜,属于膜内在蛋白。这两个预测结果相互支持。目前已知的其他植物的AMT蛋白都是位于质膜上[29],预测结果与其相符。生物信息学手段的分析预测具有一定的参考性,能够为基因功能的实验验证打下基础。
本文以拼接Uni4和Uni5为例,介绍了一种从转录组中拼接不完整基因的简易方法,显著减少了时间和成本的投入。该方法所需要的条件:1)注释为目的基因的不完整序列有多条;2)两个以上转录组库,并能够量化基因表达水平,做表达模式分析;3)序列定位于转录本5ʹ或3ʹ端且相互间没有交叠区域,有可能组装成一个转录本。
该方法源自转录组分析时的偶然发现,在盐角草转录组几个AMT基因中进行了验证,其敏感性和特异性仍需要更多的Unigene拼接试验去检验。尽管如此,该方法的提出为转录组数以万计Unigene的拼接提供了新思路,特别是对于获得基因完整编码区大有益处。并且该方法投入成本低,十分简便和快速,而且易操作,不需要复杂的生物信息学分析,应当优先考虑使用。随着测序成本的不断降低,RNA-seq技术将更加普及,所获得的序列信息也将更加丰富,借助于新技术新方法,基因克隆将变得更加简单,基因功能的研究也将更加快速。
REFERENCES
[1]Zhang K,Zhang DY,Wang L,et al.Biological features of Salicornia europaea L.and the effect of environmental factors under natural habitats in Xinjiang.Arid Land Geogr,2007,30(6):832–838 (in Chinese).张科,张道远,王雷,等.自然生境下盐角草的生物学特征及其影响因子.干旱区地理,2007, 30(6):832–838.
[2]Tikhom irova NA,Ushakova SA,Kudenko YA,et al.Potential of salt-accumulating and salt-secreting halophytic plants for recycling sodium chloride in human urine in bioregenerative life support systems.Adv Space Res,2011,48(2):378–382.
[3]Wang JP,Tian CY.Effects of N fertilization on grow th,m ineral ash absorption and accumulation of Salicornia europaea L.Agri Res Arid Areas, 2011,29(1):102–107(in Chinese).王界平,田长彦.氮肥对盐角草生长及矿质灰分累积的影响.干旱地区农业研究,2011, 29(001):102–107.
[4]Webb JM,Quinta R,Papadim itriou S,et al. Halophyte filter beds for treatment of saline wastewater from aquaculture.Water Res,2012, 46(16):5102–5114.
[5]Chen XY,Han HP,Jiang P,et al.Transformation ofβ-lycopene cyclase genes from Salicornia europaea and Arabidopsis conferred salt tolerance in Arabidopsis and tobacco.Plant Cell Physiol, 2011,52(5):909–921.
[6]Yang XL,Ji J,Wang G,et al.Over-expressing Salicornia europaea(SeNHX1)gene in tobacco improves tolerance to salt.A fr J Biotechnol,2011, 10(73):16452–16460.
[7]Tsay YF,Hsu PK.The Plant Plasma Membrane. Berlin Heidelberg:Springer-verlag,2011: 223–236.
[8]Ho CH,Tsay YF.Nitrate,ammonium,and potassium sensing and signaling.Curr Opin Plant Biol,2010,13(5):604–610.
[9]Li BZ,Merrick M,Li SM,et al.Molecular basis and regulation of ammonium transporter in rice. Rice Sci,2009,16(4):314–322.
[10]M a JB,Zhang MR,Xiao XL,et al.Global transcriptome profiling of Salicornia europaea L. shoots under NaCl treatment.PLoS ONE,2013, 8(6):e65877.
[11]Lockhart DJ,W inzeler EA.Genom ics,gene expression and DNA arrays.Nature,2000, 405(6788):827–836.
[12]Wang Z,Gerstein M,Snyder M.RNA-Seq:a revolutionary tool for transcriptom ics.Nat Rev Genet,2009,10(1):57–63.
[13]Marioni JC,Mason CE,Mane SM,et al.RNA-seq: an assessment of technical reproducibility and comparison w ith gene expression arrays.Genome Res,2008,18(9):1509–1517.
[14]Grabherr MG,Haas BJ,Yassour M,et al. Full-length transcriptome assembly from RNA-Seq data w ithout a reference genome.Nat Biotechnol,2011,29(7):644–652.
[15]M arguerat S,Bähler J.RNA-seq:from technology to biology.Cell Mol Life Sci,2010,67(4): 569–579.
[16]Wang DD,Zhu YM,Li Y,et al.Application of in silico cloning technique in plant gene engineering. J Northeast Agri Univ,2006,37(3):403–408(in Chinese).王冬冬,朱延明,李勇,等.电子克隆技术及其在植物基因工程中的应用.东北农业大学学报, 2006,37(3):403–408.
[17]Huang J,Wang JF,Zhang HS,et al.In silico cloning of glucose-6-phosphate dehydrogenase cDNA from rice(Oryza sativa L.).Acta Genet Sin, 2002,29(11):1012–1016.
[18]Chenchik A,Diachenko L,M oqadam F,et al. Full-length cDNA cloning and determ ination of mRNA 5'and 3'ends by amplification of adaptor-ligated cDNA.Biotechniques,1996,21(3): 526–534.
[19]Robertson G,Schein J,Chiu R,et al.De novo assembly and analysis of RNA-seq data.Nat M ethods,2010,7(11):909–912.
[20]Mortazavi A,Williams BA,M cCue K,et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq.Nat Methods,2008,5(7):621–628.
[21]Huang XQ,Madan A.CAP3:A DNA sequence assembly program.Genome Res,1999,9(9): 868–877.
[22]Kozak M.An analysis of 5ʹ-noncoding sequences from 699 vertebrate messenger RNAs.Nucl Acid Res,1987,15(20):8125–8148.
[23]Row ley JW,Oler AJ,Tolley ND,et al. Genome-w ide RNA-seq analysis of human and mouse platelet transcriptomes.Blood,2011, 118(14):e101–e111.
[24]Liu HL,Zheng LM,Liu QQ,et al.Studies on thetranscriptomes of non-model organisms.Hereditas, 2013,35(8):955–970(in Chinese).刘红亮,郑丽明,刘青青,等.非模式生物转录组研究.遗传,2013,35(8):955–970.
[25]Schliesky S,Gow ik U,Weber AP,et al.RNA-seq assembly–are we there yet?Front Plant Sci,2012, 3:220.
[26]Zheng L,Kostrewa D,Bernèche S,et al.The mechanism of ammonia transport based on the crystal structure of Am tB of Escherichia coli.Proc Natl Acad Sci USA,2004,101(49):17090–17095.
[27]Loque D,Lalonde S,Looger L,et al.A cytosolic trans-activation domain essential for ammonium uptake.Nature,2007,446(7132):195–198.
[28]Dyrløv Bendtsen J,Nielsen H,von Heijne G,et al. Improved prediction of signal peptides:signalP 3.0.J M ol Biol,2004,340(4):783–795.
[29]Ludew ig U,Neuhäuser B,Dynowski M. Molecular mechanisms of ammonium transport and accumulation in plants.FEBS Lett,2007, 581(12):2301–2308.
(本文责编 郝丽芳)
Assembling of an ammonium transporter gene in Salicornia europaea by expression pattern analysis of Unigene in transcriptome
Xinlong Xiao1,2,Xuan Zhang1,2,Xiaomeng W u1,2,Jinbiao M a1,and Yin’an Yao1
1 Key Laboratory of Biogeography and Bioresource in Arid Land,Xinjiang Institute of Ecology and Geography,Chinese Academy of Sciences,Urumqi 830011,Xinjiang,China 2 University of Chinese Academy of Sciences,Beijing 100049,China
RNA-seq can help us quickly obtain the whole transcriptome sequences of species under different conditions. Many Unigenes that are assembled by raw reads always do not contain complete open reading frame(ORF).In addition,it also has some redundancy in transcriptome library.Some Unigenes in the library,although belong to one transcript,cannot be assembled w ithout overlapping.We found five incomplete Unigenes annotated ammonium transporter(AMT)from Salicornia europaea transcriptome,in which two Unigenes(Uni4 and Uni5)had identical expression patterns across four transcriptomes.The two Unigenes may come from one transcript.Analyzing the Unigene position of transcript by NCBI blastx,we found that Uni4 and Uni5 respectively located in 5′end and 3′end compared w ith the reference transcript,and an unknown gap of 120 bp may exist in a hypothetic transcript to which Uni4 and Uni5 both belong.To verify the hypothesis, single forward primer and single reverse primers were respectively designed on Uni4 and Uni5,and a fragment w ith about 800 bp was generated by PCR.Then it was sequenced and aligned w ith Uni4 and Uni5.Finally,we assembled a sequence w ith 1 667 bp,which contains a complete ORF(1 482 bp,coding 494 am ino acids).It belongs to am t1 subfam ily and was named Seamt1 via the phylogenetic analysis.It was pointed by bioinformatics tools that SeAMT1 protein conformed to the AMT characteristics of other species.This work clustered expression pattern to explore the Unigenes of one transcript,and the feasibility of this method was validated through the other two groups of Unigenes.The handy method w ill benefit extension and assembling of Unigene in transcriptome,it also helps achieve the complete ORF and gene function.
RNA-seq,gene expression,sequence assembly,cloning method,RPKM,nitrogen uptake
February 26,2014;Accep ted:July 8,2014
Yin’an Yao.Tel:+86-991-7823164;E-mail:yaoya@ms.xjb.ac.cn
肖薪龙,张选,吴晓朦,等.利用分析Unigene在转录组中表达模式的方法拼接盐角草铵转运基因.生物工程学报, 2014,30(11):1763–1774.
Xiao XL,Zhang X,Wu XM,et al.Assembling of an ammonium transporter gene in Salicornia europaea by expression pattern analysis of Unigene in transcriptome.Chin J Biotech,2014,30(11):1763–1774.
Suppo rted by:National Natural Science Foundation in China(No.31270660),the Outstanding Youth Talent Foundation for Science and Technology in Xinjiang Uygur Autonomous Region of China(No.2013711018).
国家自然科学基金(No.31270660),新疆杰出青年科技人才培养项目(No.2013711018)资助。
