层状富锂锰基正极材料包覆改性的最新进展
2024-02-22李海瑶
张 建,李海瑶,梁 晨,黄 惠,高 超✉
1) 昆明理工大学冶金与能源工程学院,昆明 650093
2) 云南省冶金电极材料工程技术研究中心,昆明 650106
3) 昆明理工大学材料科学与工程学院,昆明 650093
锂离子电池(Li-ion batteries,LIBs)具有高能量密度、高功率、循环寿命长、体积小和重量轻等特点[1],近年来,电子科技及电动汽车等领域快速发展对锂离子电池的比容量、能量密度、循环稳定性等方面提出了更高要求.正极材料作为锂离子电池的重要组成部分,和锂离子电池的能量密度密切相关(占锂离子电池总成本的42%,负极约占12%).传统正极材料主要包括[2]:钴酸锂(LiCoO2)、镍酸锂(LiNiO2)、锰酸锂(LiMn2O4)、磷酸铁锂(LiFePO4)、锂镍钴锰氧化物(LiNi1-x-yMnxCoyO2,NCM)和锂镍钴铝氧化物(LiNi1-x-yAlxCoyO2,NCA).然而,这些正极材料由于较低的放电电压和比容量难以满足电动汽车(EV)和混合动力汽车(HEV)等领域对锂离子电池高能量密度的需求.
层状富锂正极材料(LMCM)具有成本低、比容量高(>300 mA·h·g-1)、能量密度高等优点,得到了国内外研究学者的广泛关注,并被视为极具发展前景的正极材料.然而,该材料工作过程中电极材料与电解液之间的界面反应及金属离子溶出等问题,导致的材料较差的循环稳定性,进而制约了其商业化应用.
层状富锂材料(xLi2MnO3·(1-x)LiMO2)是锰基电极材料重要的衍生物之一,该材料具有高比容量和低成本特点,是一种较具有研究前景的正极材料.富锂锰基材料具有独特的阴离子氧化还原机制,该机制不仅贡献了高比容量,也造成富锂材料晶体结构不稳定、氧损失和过渡金属迁移等,进而引起循环过程电压和容量衰减、较低的首圈库仑效率和较差的循环性能等问题[3-4].目前,国内外研究者通常采用表面修饰、体相掺杂、形貌结构等改性方法缓解富锂锰基材料循环过程中不可逆氧流失和相变等问题.
在众多改性方法中,表面包覆能够有效缓解富锂锰基材料表面晶格氧缺失和电解液与电极材料之间的界面反应,包覆层可以物理阻隔正极材料和电解液,防止材料表面高价金属阳离子和活性氧物质与电解液反应,及电解液分解产物腐蚀电极材料.此外,包覆层还能将充电生成的活性氧物种限制在材料内部,防止氧逃逸[5],进而缓解富锂锰基材料首圈库仑效率低、倍率性能及循环稳定性差等问题.近年来,国内外研究者在富锂锰基材料改性领域做了大量的研究工作,也发表了相关的科技论文和综述性论文.例如,Cui 等[6]和Yang等[7]发表的综述均从机理上阐述了LMCM 存在的问题,然而关于LMCM 表面包覆改性领域的系统性、多角度的报道及最新文献总结较少,影响了研究者对该领域的认知及包覆改性的应用和发展.因此,本文从包覆改性作用机理的角度出发,总结概述了前期研究者在常规包覆、双层包覆和原位包覆等方面的研究工作,并在此基础上展望了富锂锰基材料未来发展趋势,为包覆改性的商业化应用提供一定的理论基础和支撑.
1 层状富锂锰基材料的结构与问题
1.1 LMCM 的结构
富锂锰基材料的表达形式主要有以下三种,Li(1+x)M(1-x)O2、xLi2MnO3·(1-x)LiMO2和xLi(Li1/3Mn2/3)O2·(1-x)LiMO2,其中,xLi2MnO3·(1-x)LiMO2(M=Co,Ni,Mn) 是常用的一种表达形式[8].Li2MnO3和LiCoO2具有同样的层状结构,是从LiTMO2的α-NaFeO2结构衍生而来,是最典型的富锂层状氧化物,但是Li2MnO3具有Li、Mn 原子的超结构有序性,使其晶格对称性有所降低,故Li2MnO3属于单斜C2/m 空间群[9].Li2MnO3也可以写为Li(Li1/3Mn2/3)O2形式,表明TM(过渡金属)层中三分之一的位点被锂离子取代,形成了蜂窝状结构(如图1(a)所示),在该结构中,Li 和TM 离子占据由氧原子形成的交替立方晶格上的八面体位置,从[100]晶向观察的LiTMO2相和Li2MnO3相晶体结构如图1(b)所示.

图1 (a) xLi2MnO3·(1-x)LiMO2(M=Mn,Ni,Co,etc.,0<x<1) 固溶体材料的晶体学模型,具有 对称性和C2/m 对称性[6];(b) 从[100]晶向观察的LiTMO2 相(空间群:,TM=Ni、Co 和 Mn)和单斜 Li2MnO3 相(空间群:C2/m)的晶体结构[10]Fig.1 (a) xLi2MnO3·(1-x) LiMO2 (M=Mn,Ni,Co,etc.,0<x<1)crystallographic model of the solid solution material with and C2/m symmetry;(b) crystal structure of the trigonal LiTMO2 phase (space group: ,TM=Ni,Co,and Mn) and the monoclinic Li2MnO3 phase(space group: C2/m) viewed from [100] the crystallographic direction[10]
目前,关于富锂锰基材料的晶体结构仍然存在一些争议.富锂锰基材料是基于单斜晶系Li2MnO3相和菱方晶系LiMO2相的结构,一些研究者认为Li2MnO3相和LiMO2相在材料中以固溶体形式存在,而一些研究者认为富锂锰基材料是由两相组成的复合物[11-16].其中,支撑富锂锰基材料为固溶体结构的观点:一是主体结构为空间群,多余的锂离子占据过渡金属离子点位[17],二是固溶体结构为C2/m 空间群,TM 离子部分加入单斜Li2MnO3相的Li 和Mn 点位[18],该观点可以帮助理解氧和过渡金属离子的化学环境.Thackeray等[19]认为富锂锰基材料的结构是纳米级单斜晶系(C2/m)和菱面体晶系()的复合物.
关于富锂锰基材料是固溶体或两相结构的争论,有两点值得思考的地方.一是材料的组分,常见富锂材料 0.5Li2MnO3·0.5Li Mn0.5Ni0.5O2中Li :TM=1.5,Li 的数量只够支撑形成一半的Li2MnO3,而完全有序的C2/m 结构(即蜂窝状LiMn6)需要Li :TM=2 的比例才能形成.从这个角度讲,富锂材料必然不是LiMn6完全有序的固溶体结构,而是纳米尺度上Li2MnO3与LiMO2不均匀分布的嵌合体.二是富锂材料的合成条件是否有利于元素均匀分布.例如文献[11-13]采用一步混合锂源和过渡金属盐,使用熔盐或溶胶凝胶法合成材料,结果表明利用该方法制备的富锂材料是固溶体结构.而文献[14-16]采用前驱体与锂源的固相反应,结果表明所制备的材料是Li2MnO3相和菱方晶系LiMO2相两相结构.然而,富锂锰基材料的结构易受组分和合成条件的影响,基于这些差异,每种结构仅代表一种特定状态,并不适用于所有富锂锰基材料的结构.
1.2 LMCM 存在的问题
晶格氧的释放,导致较低的库仑效率的同时使得TM-O 键能降低,为过渡金属的迁移和相转变(层状→尖晶石→岩盐相)提供动力.充电过程中,锂离子从锂层脱出,部分过渡金属离子从过渡金属层迁移到锂层,Li+离子回嵌难度增大导致较低的库仑效率/倍率性能,过渡金属迁移引起的不可逆相变导致电压衰减.相变最初发生在LMCM颗粒表面,随着界面反应的发生在接下来的循环过程中造成过渡金属溶出,最终导致颗粒产生裂纹、电解液渗入颗粒内部,颗粒整体层状结构被破坏,材料的循环性能因此而急剧下降.另外,溶出的TM 离子在颗粒表面的还原/沉积增加了电池阻抗、阻碍锂离子的传输,倍率性能也因此恶化.上述这些问题,互相之间有密切联系,材料自身结构变化与材料电化学性能之间的关系,如图2 所示.

图2 富锂锰基材料结构变化与电化学性能的关系Fig.2 Structural changes and electrochemical properties of Li-rich manganese-based materials
1.2.1 首次库仑效率较差
基于充放电机理分析,富锂锰基材料的高容量特性得益于氧参与电极反应.然而,氧的氧化还原分为两部分:颗粒内部的可逆部分和表面的不可逆部分(主要是Li2MnO3组分).脱出理论为晶格中Li+的和O2-同时脱出,反应生成Li2O,不仅减少了可逆Li+,并且该Li2O 堆积在电极表面进一步抑制了Li+离子传输,导致放电过程中Li+回嵌难度增大.表面氧部分被过度氧化在初始充放电循环以O2、CO2、CO 等形式损失(如图3(a)),从而导致材料较低的初始库仑效率[20-21].Yabuuchi等[22]通过同步辐射X 光衍射,证实富锂锰材料Li1.2Mn0.54Ni0.13Co0.13O2充放电过程中存在氧和Li+同时消除现象,并且在充电末期约有7.5%的氧消除,间接证明氧脱出理论的准确性.

图3 (a~d) LMCM 结构衰退示意图[20];(e) 充放电循环过程中材料变化的示意图[29]Fig.3 (a-d) Schematic diagram of structural degradation on the surface of the LMCM material[20];(e) changes during charging-discharge cycles[29]
1.2.2 循环过程中电压/比容量衰减
Yu 等[23]利用X 射线吸收光谱技术揭示了脱锂动力学特征,与Ni 和Co 元素的更快的反应动力学相比,Mn 元素相关过程(主要是Li2MnO3组分)的脱锂动力学更差.LMCM 能带结构中氧的2p轨道和过渡金属3d 轨道的重叠导致TM—O 键表现出金属配体的特性.晶格氧的脱出,使得TM—O键能降低,也为过渡金属离子迁移提供动力[24-25].过渡金属离子迁移导致 Li+/Ni2+(TM)混排,TM 离子迁移至Li 层会堵塞Li+扩散通道,导致锂离子扩散动力学变差,同时也降低Ni 扩散动力,造成一定量容量损失和循环过程中相变(如图3(b)).Hu等[26]采用X 射线光谱和三维电子显微成像技术,通过从表面到整体,从单个粒子到整个粒子集合体,探测了富锂锰基正极材料中氧化还原电对的演变过程,研究结果表明,过渡金属阳离子的平均价态不断降低.金属原子核外层电子是费米电子,费米能级越高金属越容易失去电子,价态越低.其在报道中也阐述了富锂锰基材料循环过程中电压衰减是由于材料表面氧的析出所引起的,材料表面氧的析出导致过渡金属元素价态降低,如Mn3+到Mn2+/Co3+到Co2+,正极的费米能级升高,导致循环过程中材料的电压衰减.
1.2.3 较差的倍率性能和循环稳定性
富锂锰基材料初始充电过程中有两个电压平台,其中,低于4.5 V 的电压平台归因于LiMO2相中过渡金属离子的氧化还原反应,此时,Li+从 LiMO2组分中脱出伴随 Ni2+、Co3+的氧化反应(Mn4+价态不变).Li2MnO3表面氧的氧化还原发生在4.5 V 以上的平台,当充电电压高于4.6 V 时,超晶格Li2MnO3相开始消失,同时伴随着电解液的分解,过渡金属离子溶出及界面副反应的发生,如图3(c)和图3(d)所示.电解液分解产物HF,进一步腐蚀正极材料表面,所生成的物质附着在材料表面形成固态电解质膜(SEI),消耗锂离子,导致材料比容量损失.如图3(e)所示,TM 离子溶出将导致正极材料晶体结构坍塌,进而导致材料较差的倍率性能和循环稳定性.同时溶解的 Mn 以副产物形式在正极表面沉积,引起电极极化及电池内阻的增大,导致电压衰减和材料循环稳定性降低[27].此外,高电压下副反应产生气体的累积效应,也会导致电池的安全隐患[28].
综上所述,富锂锰基材料具有独特的阴离子氧化还原机制,该机制不仅贡献了高比容量,也造成富锂材料晶体结构不稳定,氧损失和过渡金属迁移现象明显,循环过程结构变化最终导致的电压和容量衰减等问题.
2 层状富锂锰基材料包覆改性现状
国内外研究者通常采用表面修饰、体相掺杂、形貌结构等方法缓解富锂锰基材料循环过程中晶格氧的损失和金属离子迁移,进而改善材料的循环性能和首次库仑效率等性能.在众多改性方法中,表面包覆是抑制氧流失和表面副反应的有效手段,包覆层可以物理阻隔正极材料和电解液,防止材料表面高价金属阳离子和活性氧物种与电解液反应,也能抑制电解液中HF 的侵蚀.此外,包覆层还能将充电生成的活性氧物种限制在材料内部,防止氧逃逸[30],进而缓解富锂锰基材料首圈库仑效率低、倍率性能及循环稳定性差等问题.
表面包覆主要分为物理包覆和化学包覆,包覆层主要包括电化学活性材料、非电化学活性材料和导电聚合物等,如图4 所示.电化学活性材料指既能参与循环也能够单独作为正极材料的物质,如橄榄石结构LiFePO4、LiMn2-xNixO4等,该物质在材料表面可以为正极材料 提供锂离子传输通道,进而改善材料的倍率性能.锂离子导体材料具有较高的离子导电率,有助于Li+扩散并降低循环时的电极极化.非电化学活性材料主要包括金属氧化物和金属氟化物,该包覆层能够降低表面层与HF 的反应活性,从而提高材料在循环过程中晶体结构稳定性[31].导电聚合物,如聚苯胺、聚吡咯和聚3,4-乙烯二氧噻吩/聚苯乙烯磺酸盐复合材料(PEDOT:PSS)等具有较高的电子电导率,与LMCM结合获得具有优良性能的聚合物复合电极材料,各种包覆物质的优缺点如表1 所示.包覆物质种类较多,理想的包覆材料应具有以下特征:良好的电化学稳定性、耐电解液腐蚀和在原子水平上与主体材料的晶格匹配良好,良好的离子导电性和电子导电性[32].
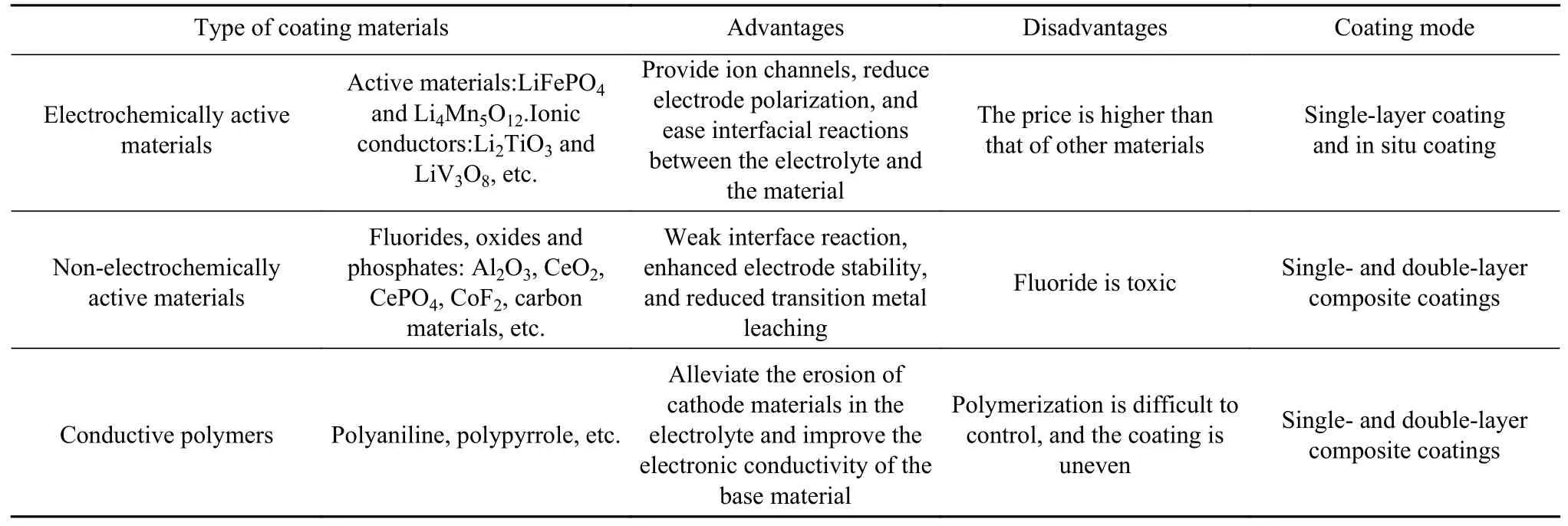
表1 不同包覆材料的优缺点Table 1 Advantages and disadvantages of different coating materials
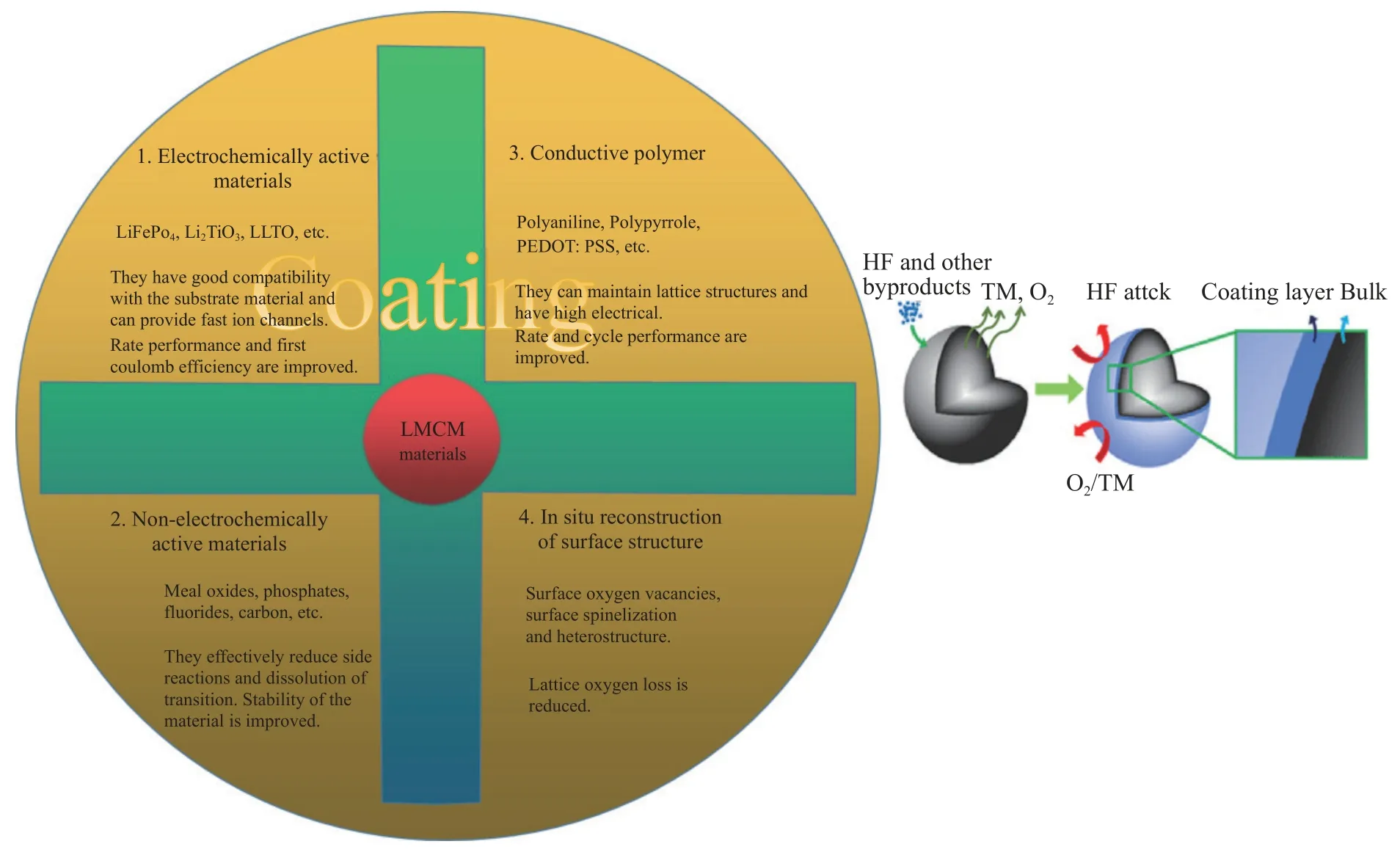
图4 常用的表面包覆物质和方法和包覆结构示意图Fig.4 Commonly used surface coating materials and methods and a schematic of the coating structure.
2.1 电化学活性材料
电化学活性材料指能够参与循环并单独作为正极材料的物质,如橄榄石结构的LiFePO4等,这种包覆物质不仅能够缓解电极材料与电解液之间的界面反应,也能为材料表面提供快速锂离子扩散通道,进而改善材料的倍率性能和循环稳定性.
Zheng 等[33]采用溶胶凝胶法制备了LiFePO4(LFP)-Li1.2Mn0.54Ni0.13Co0.13O2复合材料.图5(a)为该材料SEM 微观形貌图,图5(b)为(a)区域对应的元素分布图,从图中可以看出所有元素均匀分布在材料表面未出现明显的聚集现象,这也与图5(c)~图5(f)对应的点扫元素分析结果相一致.纳米级厚度LFP 包覆层呈薄层状均匀地包覆在正极材料LMCM 表面,如图5(g)所示.根据图5(h)和图5(i),相较于原始样品,复合材料表现出较好的电化学性能,当包覆量为5%(质量分数)时,复合材料表现出最佳的电化学性能和较小的电压衰减.在0.1C放电倍率下复合材料的容量为282.8 mA·h·g-1,比原始样品提高约80 mA·h·g-1,0.5C 下110 次(110 th)循环后为比容量249.8 mA·h·g-1,高出原始样品约100 mA·h·g-1.复合材料较好的电化学性能主要归因于,LMCM 材料表面包覆层缓解了正极材料与电解液之间的界面反应,提高了LMCM 晶体结构稳定性.此外,纳米级LFP 包覆层也为LMCM 表面提供快速的锂离子传输通道,促进锂离子的传输,进而改善材料的电化学性能.
Yu 等[34]通过Li4Mn5O12/碳纳米管材料包覆处理提高了Li1.2Mn0.54Ni0.13Co0.13O2正极材料在循环过程中的放电比容量和循环稳定性,如0.1C 放电倍率下复合材料的比容量为290.1 mA·h·g-1,10C放电倍率下比容量为183.9 mA·h·g-1,0.5C 充放电倍率下200 次循环后复合材料的容量保持率高达94.4%.Deng 等[35]通过溶剂热法制备了分级微/纳米结构和层状/尖晶石结构富锂材料,这种独特的结构结合了多孔结构和内置尖晶石隧道的优势,既可以提供三维(3D)Li+扩散通道,又可以提供空的16c 八面体位点.因此,该材料表现出较高的可逆容量(0.2C 放电倍率下比容量为302 mA·h·g-1)、较高的首次库仑效率(0.2C 放电倍率下库仑效率高达94%)和卓越的倍率性能(10C 放电倍率下比容量为193 mA·h·g-1).
快离子导体具有较高的离子导电率,可为正极材料提供锂离子快速通道,降低循环过程中材料的极化,进而改善正极材料的倍率和循环性能,是比较理想的包覆材料.Zhao 等[36]利用Li2TiO3通过同锂化方法对富锂锰基材料进行包覆改性处理,研究表明,在100 mA·g-1的大电流密度下Li2TiO3包覆改性处理后富锂层状正极材料第100 次循环放电容量高达105 mA·h·g-1,远高于原始样品的比容量(78 mA·h·g-1).Li2TiO3包覆层还降低了电极的极化和循环过程中的电压衰减.包覆改性处理后的富锂层状氧化物材料表现出优异的倍率性能和较小的电压衰减主要归因于,快离子导体Li2TiO3包覆层在延缓界面反应的同时为材料表面提供了快速的锂离子通道.这些独特的优势提高了锂离子在电极表面的迁移速度,进一步提高了富锂材料的可逆容量和倍率性能.
Meng 等[37]研究LiV3O8包覆对 [Li0.17Ni0.17Co0.10Mn0.56]O2正极材料的影响.研究表明,复合材料表现出更高的峰强度比值(I(003)/I(104)),说明LiV3O8包覆处理能够有效缓解阳离子混排.包覆处理后复合材料的库仑效率从85.8%提高到93.7%,这主要归因于钒离子的电子轨道能量高于氧离子的2p 轨道,包覆层缓解了氧的脱出,进而提高了材料的初始库仑效率(如图6(a)).Yang 等[38]利用球磨和喷雾干燥法制备Li0.75La0.42TiO3(LLTO)-Li1.2Ni0.2Mn0.6O2复合材料,复合结构如图6(b)所示.1%LLTO 复合材料在0.1C 放电倍率下的比容量高达256 mA·h·g-1,高于原始样品及其他含量复合材料的比容量(如图6(c)所示).根据图6(d),1%LLTO 复合材料也表现出最佳的循环稳定性,如30 次循环后放电容量为244 mA·h·g-1,容量保持率为95.4%,复合材料较好的电化学性能主要归因于LLTO 层不仅改善了富锂氧化物材料的离子传输,同时也缓解了电解液与富锂材料之间的界面反应.

图6 (a)样品在2.0~4.6 V,0.05C 倍率下的循环性能和库仑效率[37];(b) 样品上的 LLTO 表面包覆层;(c) 0~2%LLTO(质量分数)包覆材料的充放电曲线;(d) 0~2% LLTO(质量分数)包覆在材料0.1C 下进行30 个循环[38]Fig.6 (a) The cyclic performance and coulombic efficiency of samples at 0.05C rate between 2.0 V and 4.6 V [37];(b) LLTO surface coating on the sample;(c) charge-discharge curves of 0-2% LLTO (mass fraction) coating;(d) 0-2% LLTO (mass fraction) coating at 0.1C of the material for 30 cycles[38]
2.2 非电化学活性材料
2.2.1 金属氧化物
金属氧化物能够有效低降低表面层与HF 的反应活性,提高正极材料在循环过程中的晶体结构稳定性,因此被广泛地用于正极材料的包覆物.如 Kobayashi 等[39]利用湿法反应以Al(NO3)3·9H2O为包覆层原材料,反应、干燥后在空气气氛中450 ℃下烧结3 h,得到Al2O3-Li[Li0.2Ni0.18Co0.03Mn0.58]O2复合材料,如图7(a)所示.Al2O3均匀地包覆在Li[Li0.2Ni0.18Co0.03Mn0.58]O2材料表面,其厚度约为5 nm.如图7(b)所示,高温烧结处理过程中在Al2O3包覆层和正极材料之间形成了一层Li-Al 氧化物的中间层,该中间层有利于提高正极材料与包覆层之间的结合力,进一步提高材料在循环过程中的晶体结构稳定性.电化学测试结果表明,2%-Al2O3(氧化铝包覆量为2%)复合材料表现出较好的电化学性能,如首次放电比容量高达310 mA·h·g-1,50 ℃下30 次循环后容量保持率约92%.
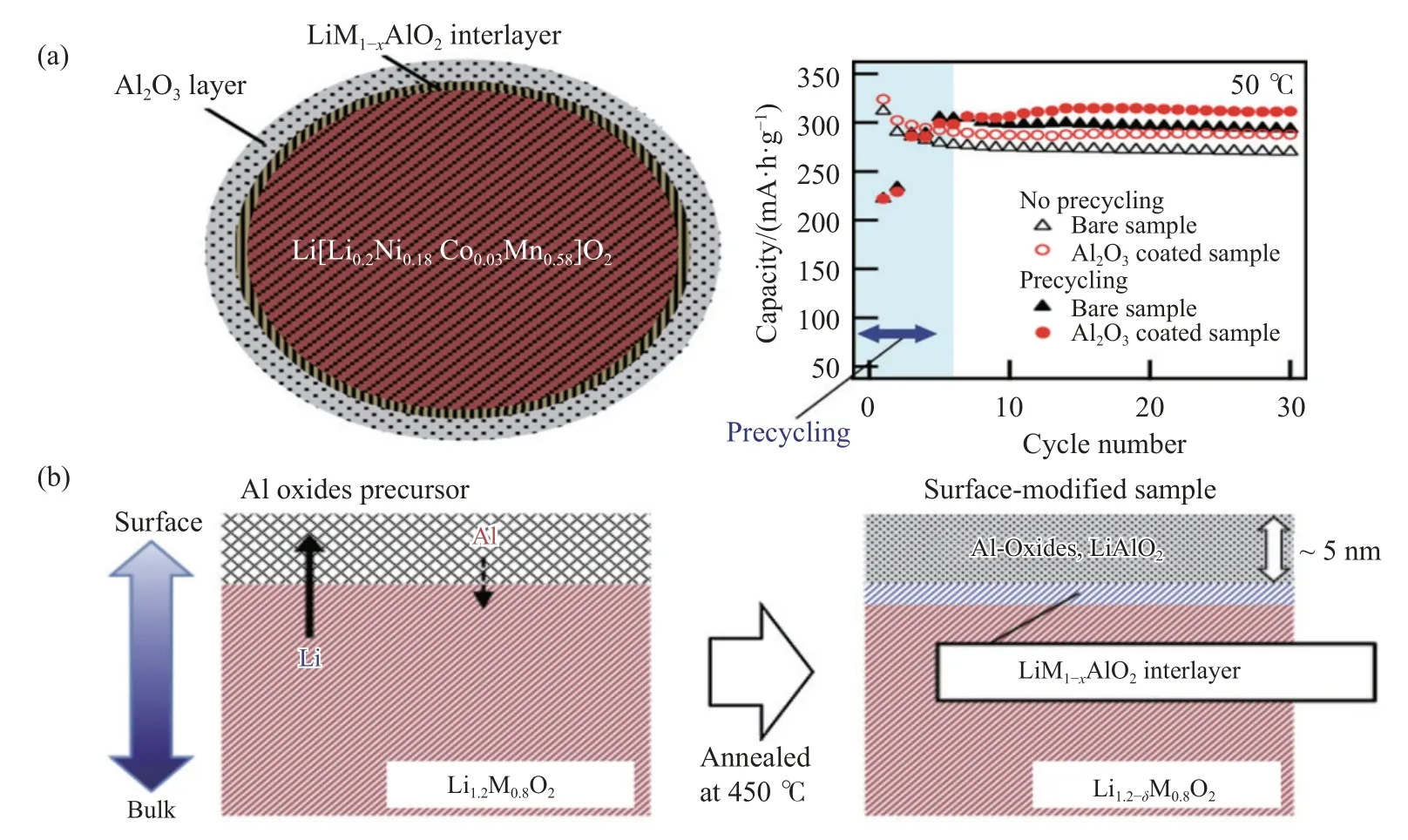
图7 (a) 合成材料的结构示意图和50 ℃下的循环性能示意图[39];(b) 2%-Al2O3 包覆层演变示意图 [39]Fig.7 (a) Schematic of the structure of the synthesized material and the cycling performance at 50 ℃ [39];(b) interpretation diagram of 2%-Al2O3 coating layer evolution [39]
Gao 等[40]等将液氮中淬火处理的Li[Li0.2Mn0.54Ni0.13Co0.13]O2与适量NH4VO3研磨混合后在空气气氛中300 ℃烧结4 h 制备Li[Li0.2Mn0.54Ni0.13Co0.13]O2-V2O5复合材料,并研究了V2O5包覆量对复合材料电化学性能的影响.研究结果表明,当包覆量为10%~12%时,复合材料表现出最大的放电比容量(0.1C 放电倍率下比容量为300 mA·h·g-1)和较高的库仑效率(约96%).复合材料电化学性能的提升机理为:包覆物V2O5可以容纳锂离子,以容纳在第一次充电后无法重新嵌入层状晶格的锂离子,进而提高材料的比容量和库仑效率.Rastgoo-Deylami 等[41]通过化学方法利用氟掺杂的TiO2(FATO)包覆改性处理Li1.2Mn0.54Ni0.13Co0.13O2材料,研究表明,包覆量为2%时复合材料表现出优异的电化学性能和较高的热稳定性,如复合材料在1C 放电倍率下的比容量高达300.1 mA·h·g-1,在10C 放电倍率下比容量为257 mA·h·g-1,在1C和5C 放电倍率下500 次循环后,容量保持率分别为91.5%和73.2%.复合材料较好的电化学性能主要归因于FATO 纳米级包覆层可以有效保护富锂颗粒表面,缓解电解液分解产物HF 对正极材料的腐蚀.此外,具有氧空位的 FATO 纳米级包覆层中氟掺杂离子进入锐钛矿结构所产生的离子导致富锂粒子在初始充电过程中释放的氧被捕获,进而提高了复合材料的库仑效率.
Yuan 等[42]通过表面化学方法对Li(Li0.17Ni0.2Co0.05Mn0.58)O2正极材料进行CeO2包覆改性处理.包覆层降低了表面电荷转移电阻,因此CeO2-Li(Li0.17Ni0.2Co0.05Mn0.58)O2复合材料表现出较大的放电比容量和较高的初始库仑效率.包覆量对复合材料的电化学性能也有较大的影响,当CeO2包覆量为1%时,改性处理后的样品表现出较高的放电比容量和较好的循环稳定性,如2 C 放电倍率下比容量为192 mA·h·g-1,远高于原始样品的比容量(172 mA·h·g-1).在0.1 C 放电倍率下循环80 次后复合材料的比容量为258.1 mA·h·g-1,远高于原始样品的比容量(229.8 mA·h·g-1),在1 C 倍率下循环80 次后容量保持率高达90.8%.复合材料较好的循环稳定性主要归因于,复合材料在脱锂状态下较好的热稳定性及CeO2纳米粒子在正极表面较好的储氧能力.
2.2.2 磷酸盐
磷酸盐包覆层能够有效地缓解正极材料与电解液之间的界面反应,并在正极表面形成稳固SEI 膜,缓解电解液分解产物对材料内部的腐蚀,进而提高正极材料的循环稳定性.如,CePO4层除了保护材料基体,还能显著提高充放电过程中正极与电解质体系的热稳定性.Chen 等[43]将层状Li1.2Ni0.13Co0.13Mn0.54O2正极与CePO4包覆层混合干燥、400 ℃下烧结5 h 在Li1.2Ni0.13Co0.13Mn0.54O2材料表面均匀包覆一层CePO4材料,并研究了包覆层对正极材料不同温度下的电化学性能的影响,研究结果表明CePO4-Li1.2Ni0.13Co0.13Mn0.54O2(CP-LNCMO)复合材料CP-LNCMO 复合材料不仅在常温下表现出较好的电化学性能在高温和低温环境下也表现出较好的电化学性能,如常温下复合材料的首次库仑效率为92.19%,远高于原始样品的首次库仑效率(88.26%),在10C 放电倍率下比容量高达110 mA·h·g-1远高于原始样品比容量(6 mA·h·g-1).55 ℃高温环境下CP-LNCMO复合材料在0.1C 放电倍率下循环20 次后,放电容为280.8 mA·h·g-1,远高于原始样品的比容量(241.1 mA·h·g-1),-20 ℃下CP-LNCMO 复合材料在0.1C 放电倍率下的比容量为246.7 mA·h·g-1,远高于原始样品的比容量(128.3 mA·h·g-1).
2.2.3 氟化物
电解液是由锂盐和多种有机溶剂混合而成,常用的配方如1 mol·L-1LiPF6的碳酸乙烯酯(EC)和碳酸二甲酯(DMC)溶液(体积比 1∶1),LiPF6对水较敏感生成HF,生成的HF 将腐蚀正极材料,导致材料较差的倍率性能和循环性能.金属氟化物能够有效缓解电极材料与电解液之间界面副反应的发生,F-也可以降低电荷转移电阻,提高正极材料的导电性和晶体结构稳定性,进而改善正极材料的倍率性能和循环稳定性.Sun 等[44]研究AlF3包覆对Li(Li0.17Ni0.2Co0.05Mn0.58)O2材料热稳定性的影响,差示扫描量热仪(DSC)研究结果表明,原始样品在241.9 ℃ 时显示出较大的放热峰,产热量为998.6 J·g-1,1% 和 2%AlF3包覆处理的复合材料在255.8 ℃时产生的热量为410.7 J·g-1,在262.5 ℃时产生的热量为273.8 J·g-1,说明AlF3包覆层能有效改善Li(Li0.17Ni0.2Co0.05Mn0.58)O2材料的热稳定性.
Chong 等[45]通过表面化学方法在Li1.2Ni0.2Mn0.6O2表面包覆一层纳米CoF2,研究表明,纳米CoF2包覆层能有效缓解首次充放电过程中O2的不可逆释放,进而改善了材料的首次库仑效率和倍率性能.如,0.1C 充放电倍率下,0.5%CoF2-Li1.2Ni0.2Mn0.6O2材料的首次放电比容量为264.4 mA·h·g-1,远高于原始样品的比容量(244.0 mA·h·g-1),5C 放电倍率下1%CoF2-Li1.2Ni0.2Mn0.6O2复合材料的比容量为167.5 mA·h·g-1,远高于原始样品的比容量(120.1 mA·h·g-1).包覆层有效改善了材料的循环稳定性,如0.1C 放电倍率下100 次循环后0.1%CoF2-Li1.2Ni0.2Mn0.6O2、0.5%CoF2-Li1.2Ni0.2Mn0.6O2和1%CoF2-Li1.2Ni0.2Mn0.6O2材料的放电容量分别为 207.2、225.3 和241.0 mA·h·g-1,容量保持率分别为 81.5%、85.2% 和 93.0%,远高于原始样品比容量(153 mA·h·g-1)和容量保持率(63%).复合材料优异的倍率性能及循环稳定性主要归因于电解质-电极反应界面的纳米包覆层缓解界面反应和TM离子溶出,缓解O2释放和循环过程中材料晶体结构从层状结构到尖晶石结构的相变.
2.2.4 碳材料
类石墨烯碳被证实具有典型的类石墨烯层状碳结构,石墨烯具有较高的电导率可以降低电池电阻,尤其是电荷转移电阻.石墨烯导电框架能有效地缓解LMCM 材料在循环过程中的极化,从而提高电池倍率性能和循环稳定性.
Jiang 等[46]使用液相混合法制备了LMNCO-G复合材料,该材料在3C 和12C 放电倍率下比容量分别为120 mA·h·g-1和50 mA·h·g-1,复合材料较好的电化学性能主要归因于石墨烯较好的电子导电性,石墨烯包覆层降低了电极材料的反应超电势,进而提高材料的倍率性能.Zhuo 等[47]等使用LPAN(液态聚丙烯腈)为碳源在Li[Li0.1Ni0.45Mn0.45]O2正极材料表面均匀包覆一层碳材料,如图8 所示.研究结果表明,当LPAN 包覆量为12%时,复合材料在0.1C 放电倍率下比容量为 210 mA·h·g-1,0.1C 放电倍率50 次循环后,容量保持率接近100%.复合材料较高的放电比容量和较好的循环稳定性主要归因于LPAN 包覆层在循环过程中能有效缓解锰离子的溶出及改善正极材料的电子导电性.Zheng等[48]通过溶胶凝胶法在层状氧化物Li1.2Mn0.54Ni0.13Co0.13O2材料表面包覆一层氟 (F) 掺杂的碳(LMCNO@CF),研究结果表明,碳材料与正极材料之间较强的电子耦合作用,有效地提高了正极材料的结构稳定性和电子传导性,进而改善了材料的倍率和循环稳定性能.

图8 以LPAN 为碳源制备层状包覆类石墨烯的Li[Li0.1Ni0.45Mn0.45]O2 的示意图[47]Fig.8 Illustration of the preparation of layered Li[Li0.1Ni0.45Mn0.45]O2 in situ coated with graphene-like carbon from LPAN[47]
2.3 导电聚合物
导电聚合物包覆层不仅能物理阻隔正极材料和电解液,防止材料表面高价金属阳离子和活性氧物种与电解液反应,抑制电解液中HF 的侵蚀正极材料,也能提高基体材料的电子导电性,因此也常被用作包覆物质.如,Wang 等[49]采用聚苯胺包覆球形LMCM(Li1.5Ni0.2Co0.2Mn0.6),研究结果表明,聚苯胺包覆层可以有效地改善球形LMCM材料的库仑效率和循环稳定性,如0.5C 放电倍率下200 次循环后容量保持率为92.4%,0.1C 和10C 放电倍率下比容量分别为302.9 mA·h·g-1和146.2 mA·h·g-1.Wu 等[50]研究了导电聚吡咯包覆层(PPy)对层状结构Li1.2Mn0.54Co0.13Ni0.13O2材料电化学性能的影响,研究表明,PPy 包覆层能够有效抑制高电位下的电解质分解和氧空位消除,进而提高材料的初始库仑效率和循环稳定性.
Wu 等[51]利用 PEDOT:PSS 复合材料在Li1.2Ni0.2Mn0.6O2颗粒表面形成了厚度为5~20 nm 无定形导电聚合物薄膜.研究结果表明,当包覆量为3%时,复合材料表现出更好的倍率性能和循环性能,在0.1C 放电倍率下,比容量为286.5 mA·h·g-1,在1C放电倍率下100 次循环后比容量为146.9 mA·h·g-1.复合材料较好的电化学性能主要归因于导电聚合物包覆层较好的电子导电性,包覆层缓解了电极材料与电解液之间的界面反应,进而改善了材料在循环过程中的晶体结构稳定性.
2.4 双层包覆改性现状
单层包覆改性虽能缓解电解液与电极材料之间的界面反应,但往往会影响电子或锂离子传输,如金属氧化物包覆能改善基体材料的电子导电性,但在一定程度上影响基体材料锂离子导电性,快离子导体虽能提高锂离子传输速率但在一定程度上阻碍电子的传输.相较于单层包覆,双包覆层一般是由离子导体层和电子导体层组成,因此,双层包覆有效解决了单层包覆不能兼顾离子和电子导电性问题.
Lu 等[52]采用溶剂热法和原位同步碳化还原法合成了尖晶石层和碳层双层包覆0.5Li2MnO3·0.5Li[Mn1/3Ni1/3Co1/3]O2的复合材料正极材料LR@S@C(LR 属于一种富锂材料,LR@S@C 为复合材料),如图9(a)所示.尖晶石结构(Fd-3m)包覆层为基体材料提供了快速Li+扩散通道,碳包覆层为基体材料提供了电子快速传输通道,因此,该复合材料表现出较好的倍率性能和循环稳定性,如0.2C放电倍率下比容量高达313.9 mA·h·g-1,5C 放电倍率下比容量为186.1 mA·h·g-1,1C 放电倍率下100 次循环后容量保持率为102.7%,如图9(b)所示.

图9 (a) LR@S@C 的界面结构[52];(b) LR 和LR@S@C 材料在1C 下的循环性能[52];(c) LMR 和LMR@Spinel@MgF2 材料在循环过程中的结构转变示意图[53]Fig.9 (a) Interface structure of LR@S@C[52];(b) cycling performance of LR and LR@S@C at 1C rate[52];(c) illustration of the structural transformation of the LMR and LMR@Spinel@MgF2 materials during cycling [53]
Zhu 等[53]通过液相浸渍沉积法在Li1.2Mn0.6Ni0.2O2(LMR,属于富锂材料的一种)表面依次包覆尖晶石结构Li4Mn5O12和非晶态MgF2,制备了双层包覆复合材料(LMR@Spinel@MgF2).富含锂尖晶石层Li4Mn5O12为正极材料提供3D 锂离子通道,并且可以缓解电极与电解质之间的副反应以及O2的释放,外层非晶MgF2包覆层有利于锂离子迁移并进一步缓解HF 对Li4Mn5O12的侵蚀,缓解了正极材料从层状结构到尖晶石型Li4Mn5O12结构转变(如图9(c)所示),因此,复合材料表现出较高的首次库仑效率(96.4%)、较好的循环稳定性(0.5C 倍率下300 次循环后容量保持率为80%)和较小的压降(0.45 V).Chen 等[54]利用AlF3和石墨烯双包覆层改性处理尺寸为100 nm 纳米富锂氧化物(Li-Rich@AlF3@Graphene),纳米富锂氧化物颗粒表面AlF3包覆层厚度为2 nm,外层用包覆层为石墨烯片.电化学研究结果表明,AlF3和石墨烯双包覆层能有效地改善富锂氧化物材料的循环稳定性和倍率性能,如2C 放电倍率下250 次循环后容量保持率接近70%,5C 倍率下放电比容量仍高于100 mA·h·g-1.复合材料较好的电化学性能主要归因于,石墨烯包覆层较好的电子导电性促进了电极和电解质之间界面上的电荷转移,提高材料的倍率性能.此外,AlF3包覆层也能有效地缓解电解液分解产物对富锂氧化物的的侵蚀,减少溶解过渡金属离子溶出,进而提高材料在循环过程中的晶体结构稳定性和循环稳定性.
2.5 表面原位包覆现状
根据文献报道,在富锂材料表面通过气固界面反应原位诱导形成氧空位层[55-57]、生成阳离子无序结构相和强P—O 键稳定表面晶格氧[58],进一步缓解TM 离子的溶解.表面进行尖晶石化处理,形成3D 结构的尖晶石相层,可以有效提高Li+扩散速率.表面引入岩盐相层,有助于缓解氧气的释放,可逆氧化得到改善,提高材料的稳定性.这些表面原位化学“包覆”处理手段类似于化学镀,保证包覆层和本体结构的结构完整性和连续性,所形成的包覆层具有良好的化学附着性能和机械性能.
2.5.1 表面气体处理
LMCM 的高容量来源于晶格中的氧参与氧化还原,当Li+和O 2p 不稳定电子从Li—O—Li 构型中脱出,相邻的氧原子将聚合并形成 O—O 二聚体以补偿电荷.在这种情况下,一些O—O 二聚体会过度氧化并以O2的形式通过表面释放,出现不可逆阴离子氧化还原,导致材料较低的首次库仑效率.为此,研究者将氧空位(Oxygen vacancies,OVs)引入LMCM 表面,即在合成过程中提取材料表面上部分晶格氧而不引起结构破坏,进而抑制氧气的释放和提高电化学性能.通过PH3气体处理LMCM 表面生成阳离子无序结构相和强P—O 键稳定表面晶格氧,进一步缓解TM 离子的溶解.
Qiu 等[56]利用CO2与Li[Li0.144Ni0.136Co0.136Mn0.544]O2的气固界面反应(GSIR),在材料表面形成一层均匀的氧空位(OVs),如图10 所示,CO2在材料表面获取O 原子,与材料中游离锂形成-xLi2CO3产物.氧空位能够减少氧释放,进而提高首次库仑效率.通过在不影响富锂层状氧化物结构完整性的情况下,利用二氧化碳和富锂层状氧化物的摩尔比约为1∶5,在200 ℃下加热10 h,合成具有表面氧空位的富锂正极材料(GSIR-LR-NCM).作者使用差分电化学质谱以评估初始充放电过程中的气体释放,对原始样品和GSIR-LR-NCM,在4.5~4.6 V附近进行O2检测.结果显示GSIR-LR-NCM 材料释放的O2气体比原始材料少得多.较少的O2释放也有助于减少对电解质的副反应和形成较薄的SEI 层,进一步降低电极/电解质界面处的电化学电阻.OVs 对激活锁定在四面体位点的Li+有很好的作用,为Li+的扩散提供了良好的环境.该改性实现了均匀地产生氧空位来实现对氧活性的精细控制,进一步发现在20 nm 厚的表面区域中的氧空位通过激活四面体位点中 Li+,并抑制气体从表面释放来,促进Li+扩散,最终实现改性后的富锂层状氧化物具有在1C 下150 次循环后保持超过260 mA·h·g-1的放电容量,而电压也没有明显衰减.Cui 等[57]通过调控微/纳米结构,成功合成不规则的微球、微棒颗粒,研究表明,材料表面产生氧空位的含量随着形态的变化而变化.
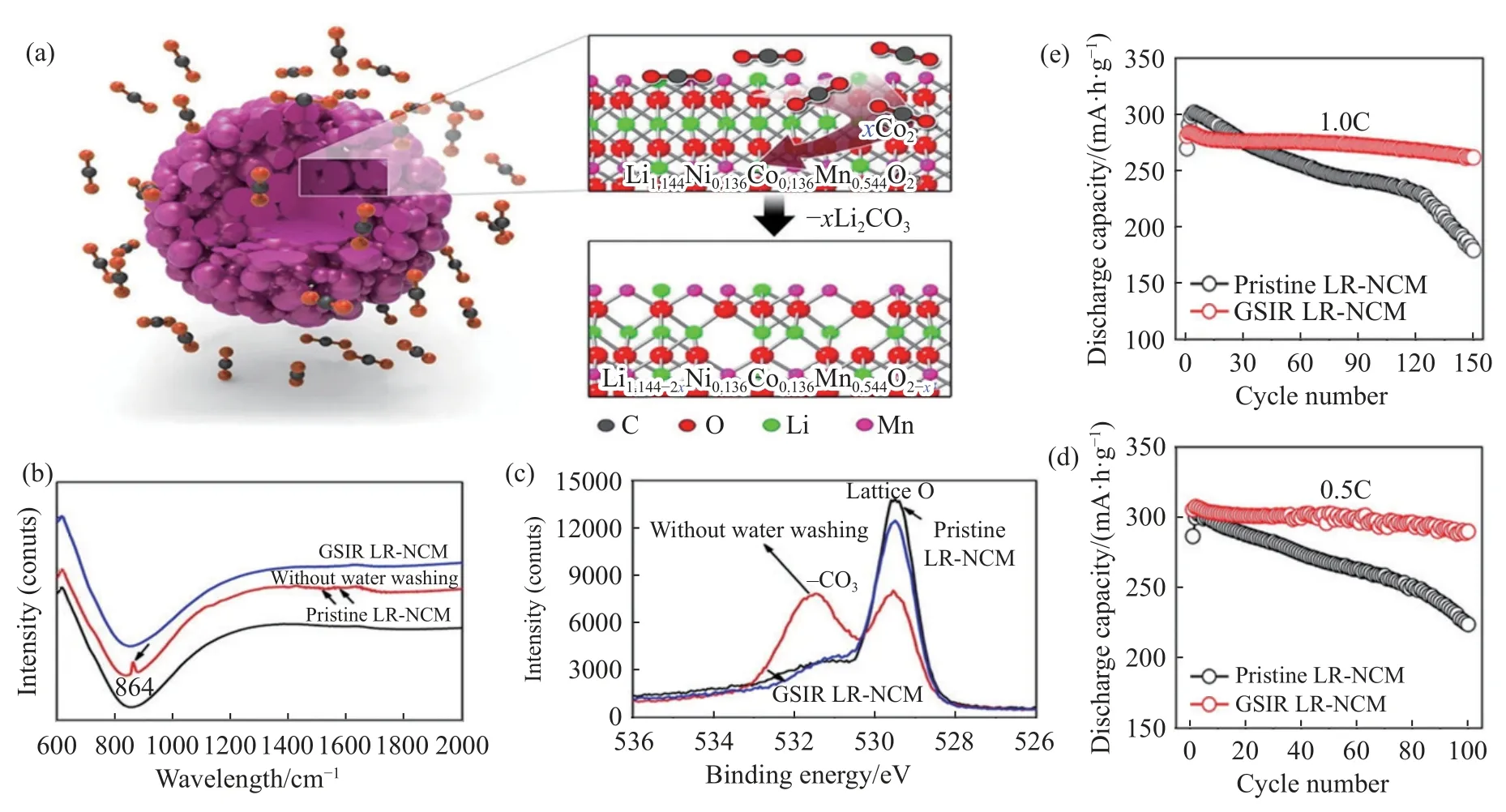
图10 (a) 富锂层状氧化物和二氧化碳之间的GSIR 示意图;(b) FTIR 光谱;(c) O 原子1s 的XPS 光谱;(d~e) 原始和GSIR LR-NCM 在0.5 C 和1.0 C,55 ℃下的循环性能[56]Fig.10 (a) Schematic of GSIR between Li-rich layered oxides and carbon dioxide;(b) FTIR spectrum;(c) XPS spectra of the O1s;(d-e) cycling performance of the pristine and GSIR LR-NCM at 55 °C,0.5C and 1.0C [56]
Sun 等[58]在低温(300 ℃)下对(Li1.17Mn0.44Ni0.34Co0.04O2)进行PH3气固处理,制备表面具有含磷复合保护层的样品(P-LNCM).如图11(a)所示,通过次磷酸钠(NaH2PO2)的热解反应得到PH3气体,如:2NaH2PO2(s)→Na2HPO4(s)+PH3(g)↑(s 为固体,g 为气体).将一定量的LNCM 样品和次磷酸钠分别装入不同坩埚中,在Ar 气氛下300 ℃加热2 h,形成了独特的LiTMPO4相和阳离子无序相.研究表明,P-LNCM 在1C 倍率条件下经过250 次循环后,比容量达到176.7 mA·h·g-1,远高于原始样品的138.1 mA·h·g-1.相当于P-LNCM 在250 次循环后的容量保留率为84.1%,LNCM 为69.9%,这也与容量微分(dQ/dV)曲线研究结果相一致,如图11(b)所示,图中等高线的强度和位置分别代表材料的放电比容量和还原峰的位置.随着循环次数的增加,LNCM 材料的峰值强度呈持续下降趋势,250次循环后,LNCM 的峰值位置从 3.6 V 降至约3.3 V,说明该材料的放电电压和比容量出现了严重的衰减现象.P-LNCM 材料的峰值位置从3.7 V 左右开始,250 次循环后位置和强度基本没有偏移,说明该材料较好的稳定性,这归结于复合保护层的阳离子无序结构相和P—O 键稳定表面晶格氧,LiTMPO4相具有抗高电位的能力,缓解电极和电解质之间的副反应,使其在高电压循环下保持稳定.
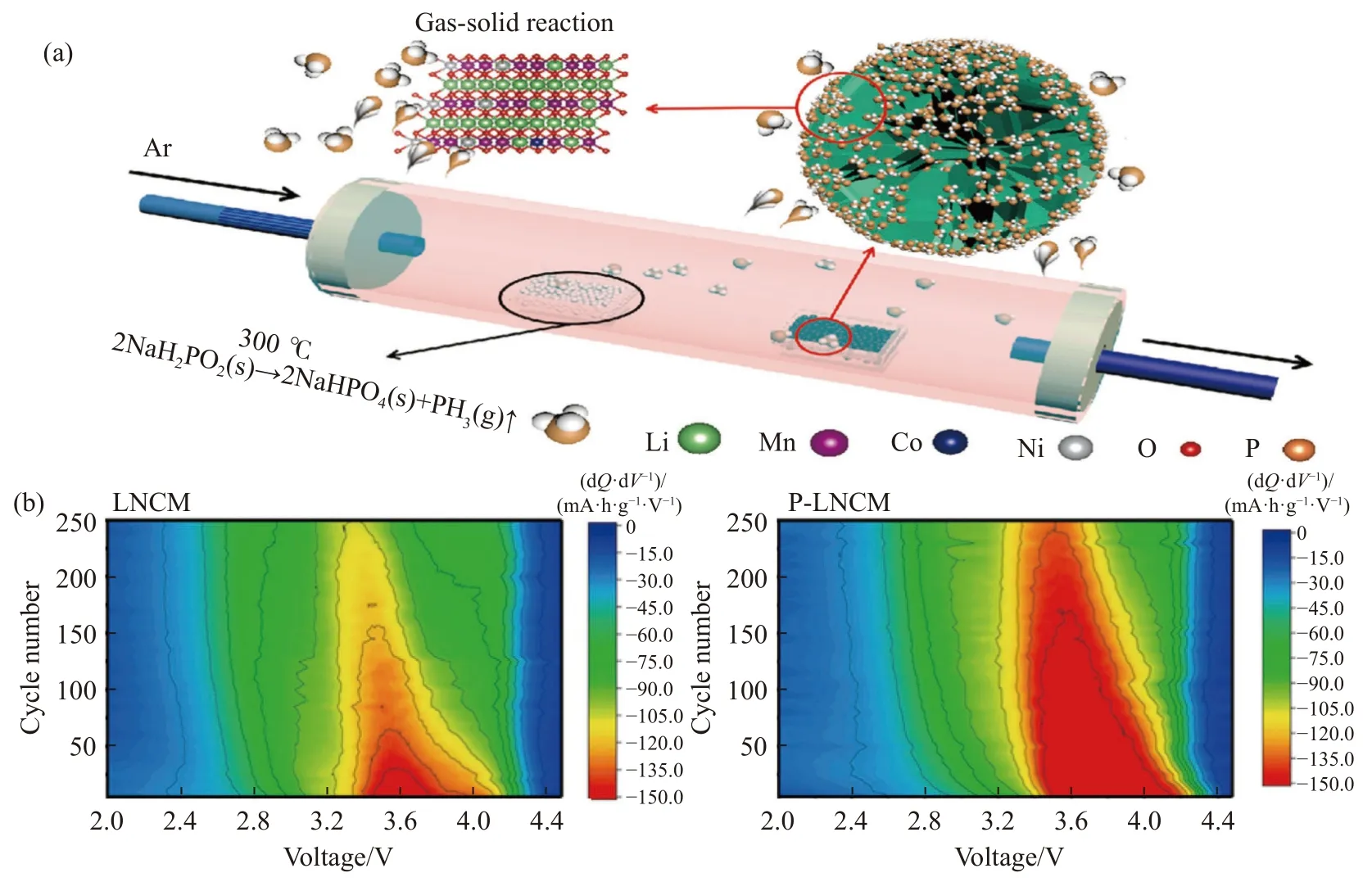
图11 (a) PH3 表面改性过程示意图;(b) LNCM 和P-LNCM 电极的微分放电容量曲线等高线图[58]Fig.11 (a) Schematic of the PH3 surface modification process;(b) the contour plots of differential discharge capacity curves for LNCM and P-LNCM electrodes [58]
2.5.2 表面尖晶石化
与层状结构的2D 骨架相比,3D 结构的尖晶石相在循环过程中能够为材料表面提供更多的Li+扩散通道,提高Li+传输速率,进而改善材料的循环稳定性和倍率性能.控制合成条件和材料表面处理实现材料表面尖晶石化的主要方法.控制合成条件主要是通过制备过程中调节煅烧温度、时间及材料的组成成分等实现富锂锰基材料表面尖晶石化,其中,通过调整锂含量实现材料表面尖晶石化是目前常用的方法.对于层状结构富锂锰基材料,Li+的迁移仅发生在层状结构的过渡金属和锂层之间,材料表面尖晶石结构能够提供更多传输位点,允许Li+通过具有连续3D 构型的四面体8a 位点扩散,促进循环过程中锂离子的传输.Jin 等[59]采用聚合阴离子诱导法,在0.5Li2MnO3·0.5LiNi0.5Mn0.5O2层状材料制备过程中加入聚偏氟乙烯(PVDF),煅烧后在复合材料基体表面上发现了部分的尖晶石相的LiNi0.5Mn1.5O4.通过结构、形态和电化学表征系统地证实尖晶石相的形成主要归因于PVDF 的氟化作用.在750 ℃的温度下制备的复合材料表现出优异的倍率性能,材料优异的倍率性能主要归因于尖晶石结构的形成.
Yu 等[60]提出一种双导电表面控制策略,通过将碳纳米管(CNT)网络经过热处理和化学浸出效应结合,将LMCM 表面诱导为CNT 骨架和尖晶石外延层的双重导电表面.由于CNT 具有较高的电子电导率和尖晶石结构材料快速的锂离子传输通道,改性处理后复合材料表现出优异的倍率性能(10C 时为195 mA·h·g-1)和较高的能量密度(0.1C时为1077 W·h·kg-1).
2.5.3 层状岩盐异质结构
与尖晶石相重建相比,层状岩盐异质结构报道较少,表面原位岩盐异质结构也能够有效低缓解循环过程中O2的释放,进而稳定LMCM 的结构.与原位尖晶石表面重建相似,原始LMCM 中岩盐表面的重建也伴随着TM 离子的富集.然而,对于TM 富集的类型,目前主要存在两种不同的看法,如Jarvis 等[61]认为岩盐相的生成层与表面Ni富集有关,Yan 等[62]认为岩盐相表面重构与表面Co 偏析有关.
Boivin 等[63]通过溶胶-凝胶法利用Ni、Co 和Ni/Co 对Li[Li0.2Ni0.13Co0.13Mn0.54]O2中Li2MO3组分进行Ni 或Co 单掺杂或者Ni/Co 共掺杂,制备了包括(LNMO)、(LMCO)和(LNMCO)三种样品.发现Ni 掺杂和Ni/Co 共掺杂使Li[Li0.2Ni0.13Co0.13Mn0.54]O2材料表面出现岩盐相,其中单掺杂Ni 时,形成的相层更致密,抑制氧损失方面效果明显.这种富镍贫锂岩盐表面相作为保护层来抑制氧气的释放,有利于可逆氧化,减少首次充电时由于O2-的氧化导致Li2MO3组分中晶格中氧的损失,从而提高库仑效率.
Cao 等[64]提出一种在富锂正极材料表面涂覆锂化过渡金属磷酸盐的方法,在LRMO (Li1.2Mn0.54Co0.13Ni0.13O2)和LiMPO4的界面生成一种异质外延纳米结构.第一步,采用NaH2PO4蚀刻MCO3(M=Ni,Co,Mn)前驱体.蚀刻步骤导致界面相转换,记为MCO3@M3(PO4)2.第二步,将MCO3@M3(PO4)2复合材料与Li2CO3混合,在800 ℃空气气氛下煅烧.在过程中MCO3@M3(PO4)2发生了共锂作用,最终形成了包覆LiMPO4的LRMO(记为LRMO@LiMPO4),如图12 所示.异质外延纳米结构促进Li+固态扩散动力学,缓解了正极材料受到的电解质腐蚀.LiMPO4的包覆导致更多的氧空位和LiMn2O4尖晶石相的形成,这两者都可以通过抑制循环过程中的氧释放有效地减少不可逆容量损失.基于上述特性,LRMO@LiMPO4正极材料表现出高倍率性能和循环稳定性:在 5C 倍率下放电容量为156 mA·h·g-1和在 0.5C 下经过140 个循环后的容量保持率为93.4%.

图12 具有异质外延结构的LRMO@ LiMPO4 的制备示意图[64]Fig.12 Schematic of the fabrication of LRMO@ LiMPO4 with a heteroepitaxial structure at the interface[64]
3 包覆改性机理探讨
表面包覆层可以缓解活性材料与电解质之间的副反应,对材料的循环性能和热稳定性也有一定的改善,已被证明是缓解LMCM 容量损失的有效方法.包覆改性的机理如下:
富锂锰基材料在高电压状态下,电解质和正极界面发生副反应.电极表面的活性氧氧化有机溶剂,释放出CO2,反应方程式(1).氧化物续腐蚀电极材料,反应方程式如式(2).另外,电解液在制备过程中含有少量水,会与LiPF6反应如方程式(3),产生HF[65-66].HF 进一步腐蚀电极材料,产生LiF 附着在材料表面,造成材料界面阻抗的增加和比容量的损失.同时,随着循环的进行,Mn 发生歧化反应,可溶解在电解液中,进一步加剧了层状结构的构型变化和结构的坍塌[67].
表面包覆改性处理缓解电解液与电极材料的接触,弱化电解质和正极界面发生副反应,减少气体释放和过渡金属迁移/溶出,进而缓解层状结构转变、提高了材料的结构稳定性、提高锂离子电池容量和电化学循环稳定性[68],示意图如图13.

图13 包覆层对材料形成保护的原理示意图[69]Fig.13 Schematic of the principle that the coating layer protects the material [69]
4 结语与展望
富锂锰基材料固有问题有:(1)阳离子氧化还原反应,阴离子氧化还原反应提供额外的容量,以及氧气释放导致颗粒表面形成微孔和阳离子价态降低,颗粒内产生微应变以及从层状到尖晶石的相变,循环中材料的膨胀率上升,最终导致较低ICE(首次库仑效率)、快速电压衰减和容量衰减.(2) LMCM 在合成过程中的材料缺陷,如Ni2+/Li+阳离子混排,表面残留锂化合物(LiOH、Li2O 和 Li2CO3)以及元素偏析也会导致动力学不良和电化学性能衰减.(3)材料固有的扩散动力学差使其难以获得高倍率性能,界面反应属于活性物质与电解质之间的问题.
因此,活性物质材料表面使用电化学活性材料、非电化学活性材料和导电聚合物等物质包覆可以缓解活性材料与电解质之间的副反应,对材料的循环性能和热稳定性也有一定的改善,已被证明是控制LMCM 容量损失的有效方法.尽管表面包覆能有效改善容量损失和循环稳定性,除了进行包覆改性外,富锂材料还需通过调整合成工艺,对材料进行晶面结构调控和掺杂等手段.来稳定晶格结构、抑制材料内部层状结构向尖晶石结构的转变,辅助解决材料内部的稳定性.通过表面结构原位重建:诱导氧空位,表面尖晶石化以及层状岩盐异质结构等,来稳定表面结构、抑制表面释氧、提高锂离子的传输的动力学和抑制电极极化.诸如此类的集成化改性是未来的趋势.
另外,LMCM 的发展还应考虑一些实际应用问题.如,当前主要通过半电池研究LMCM,难以评估在实际应用中的相关性能参数.后续全电池组装测试研究也需要重点关注,例如,选择合适的负极材料、开发耐高压(>5 V)电解质、添加耐高压添加剂和制造固态电解质等.其中,固态电解质取代液态电解质是一项革新性措施,但界面问题比较棘手,需要进一步开展研究.安全问题方面,LMCM 热失控、气体逸出和其他可能的副反应是关键因素,也需要进一步探索研究.此外,锂、镍、钴的资源短缺,以及钴的高价格,开发无钴富锂正极材料迫在眉睫.基于LMCM 的潜力,期待LMCM的商业化可以早日实现,也希望这篇综述在为表面包覆提供参考的同时能激发科技工作者对LMCM正极材料表面结构研究的浓厚兴趣,以推动下一代锂离子电池能量密度开启新篇章.
