氧离子导体表面包覆对Li2MnO3正极材料性能的影响*
2023-11-09李东林刘小九陆继承
张 龙,李东林,刘小九,陆继承
(长安大学 材料科学与工程学院,新能源材料和设备实验室,西安 710061)
0 引 言
富锂锰基正极材料,通式为xLi2MnO3·(1-x)LiMO2(M为Ni、Mn、Co等),作为一种极有前景的高性能锂离子阴极材料的替代品,因其非常高的比容量(大于250 mAh·g-1)、高能量密度和相对较低的成本而受到广泛关注[1]。但其在充放电过程中放电平台电压衰减和容量衰减阻碍了其在商业化的进一步发展。已有的研究表明,富锂锰基材料的这种衰减行为与Li2MnO3组份密切相关,且在Li2MnO3正极材料中也发现了同样的问题[2-4]。因此,研究Li2MnO3正极材料的电化学行为,有助于更好地理解富锂锰基正极材料的这种衰减行为。目前,大部分研究人员认为这种衰减行为是由于Li2MnO3中4.5V以上发生的不可逆的氧氧化还原反应所生成的氧气引起的,在循环过程中,不可逆的氧释放就跟多米诺骨牌效应一样,导致放电电压平台和容量的持续衰减[5-10]。因此,抑制Li2MnO3中不可逆的氧释放,或许可以防止这些问题的发生。Lu等[11]利用离子交换法实现对Li2MnO3的重氟化(9.5%),其表现出极好的电化学性能。在第一个循环中,完全抑制了O2的演化,极大增加了氧氧化还原反应的可逆性,实现了389 mAh·g-1的超高比容量。在50 mA·g-1的电流密度下,循环120圈还有91.8%的超高容量保持率。他们认为氟离子的引入,使得颗粒表面产生一层薄的尖晶石层,极大提高了表面结构的稳定性,使得LMOF表现出优越的循环稳定性和倍率性能;最近Wu等[12]在Li2MnO3结构中引入堆垛层错缺陷,稳定了氧氧化还原反应,消除了氧的释放,在电化学上表现出高的可逆比容量(320 mAh·g-1)和循环稳定性(100次后86%)。可见,抑制Li2MnO3中不可逆的氧释放,可以表现出更好的电化学性能。
针对这一问题,氧离子导体由于具有氧离子迁移这一特性,可以考虑作为一种改性手段,其在固体氧化物燃料电池(SOFC)方面应用较多[13]。目前,研究最多的是ZrO2基氧离子导体。通过对ZrO2进行掺杂二价或三价氧化物,使其晶格中产生大量的氧空位,从而具有氧离子导电性[14-15]。基于这样的理论,使用氧离子导体对富锂材料Li2MnO3进行包覆。或许可以减少不可逆的氧释放。借鉴单科等[16]的研究,本文选择8%的Sr对ZrO2进行掺杂(Zr0.92Sr0.08O2),再对Li2MnO3进行包覆改性。
1 实 验
1.1 材料的合成
本文使用溶胶凝胶法合成纯Li2MnO3材料,其中一水合柠檬酸跟金属离子按1∶1混合,Li源过量5%。将一水合柠檬酸倒入烧杯中,加入30 ml的去离子水,在40 ℃下水浴加热搅拌;待一水合柠檬酸溶解完后,再加入乙酸锰;待锰盐溶解后,再加入乙酸锂,在40 ℃下搅拌1.5 h;然后将氨水逐滴地加入到溶液中,使得溶液pH达到9.0,搅拌1.5 h;最后在80 ℃下对溶液进行蒸发,如图1为简单的合成工艺示意图。然后将湿凝胶放在鼓风烘箱中,在150 ℃下保温12 h,研磨处理,再在马弗炉中700 ℃保温4 h,研磨处理,再在马弗炉中700 ℃保温4 h,最后就可以得到Li2MnO3正极材料。

图1 Li2MnO3粉末制备前期合成工艺示意图Fig.1 Schematic diagram of the preliminary synthesis process of Li2MnO3 powder preparation
针对包覆材料,需要先配制Zr0.92Sr0.08O2溶液。具体来讲,将正丙醇锆、乙酰丙酮跟40g的无水乙醇混合,在40 ℃的水浴锅中进行搅拌;与此同时,将乙酸锶与10g的冰醋酸混合,也在水浴锅中40 ℃搅拌;待锆溶液变清澈后,将锶溶液逐滴加入到锆溶液中,40 ℃搅拌1h。然后取混合溶液的1%和3%,分别加入到Li2MnO3材料中。然后搅拌超声数次,在真空下保持一晚,再在80 ℃下蒸干;最后,将3种材料放在65 ℃的鼓风干燥箱中进行干燥处理,再在马弗炉中650 ℃保温3h即可。此外,为了更好地了解包覆材料的结构特性,最后还制备了8%的Zr0.92Sr0.08O2包覆Li2MnO3。为了方便起见,将包覆0%,1%,3%和8%的材料分别命名为LMO,LMO@1ZMO,LMO@3ZMO和LMO@8ZSO。
1.2 材料的表征
在本论文中,使用德国Broker公司生产的型号为AXSD8的粉末X射线衍射仪来研究晶体物质的微观结构,扫描角度2θ为10~90°范围内,扫描速度为2.6°·min-1,以Cu靶Kα为辐射源(其中λ为0.15406 nm),工作电流为40 mA,工作电压为40 kV。采用日本日立公司生产的的型号为Hitachi S-4800的扫描电子显微镜来研究材料物质的微观形貌,工作电压为15 kV,工作电流为10 μA。采用英国Kratos公司生产的型号为AXIS Ultra的X射线光电子能谱仪来研究材料微观区域的元素种类和含量。
1.3 材料的电化学性能测试
在本论文中,活性物质为改性前后的材料,导电剂采用科琴黑,将活性物质、科琴黑和PVDF在NMP中按照7:2:1的质量比混合均匀,搅拌均匀后用玻璃棒将浆料涂覆在铝箔表面上;最后将极片放置在真空干燥箱中,氩气气氛下100 ℃烘12 h即可。隔膜采用聚丙烯微孔膜,电解液采用1 mol/L的LiPF6溶解于体积比为1∶1∶1的碳酸二甲酯(DMC)、乙烯碳酸酯(EC)和碳酸甲乙酯(EMC)中,在充满氩气的手套箱中进行CR2025型扣式半电池的组装,同时用电池封口机对电池进行快速封装。采用深圳新威电池测试系统在室温下2.0~4.8 V电压范围内对电池进行充放电测试。采用型号为Versa STAT3的电化学工作站对电池进行循环伏安测试,测试电压范围为2~4.8 V,扫描速度为0.1 mV·s-1。
2 结果与讨论
2.1 材料结构分析
2.1.1 XRD分析
如图2(a),为四种样品的XRD图谱。其中,纯LMO的衍射峰与标准PDF卡片PDF#84-1634的峰位基本保持一致,表现为单斜层状结构,空间群为C2/m。其中,在2θ为20°和25°之间,可以看见一个很明显的衍射峰,这是Li2MnO3材料特殊的超晶格衍射峰,对应于过渡金属层中Li/Mn的有序排列[17]。通过观察,发现改性材料的衍射峰几乎与纯LMO保持一致,表明对材料进行包覆时不会改变原始材料的结构。而且还能直观的看到,随着包覆量的增加,整体的衍射峰变得越来越强,表明包覆量的增加会提高材料的结晶度。另外,位于(-133)和(-331)峰分裂越明显,表明材料的层状结构越好[18]。可以看出,四种材料都具有良好的层状结构。
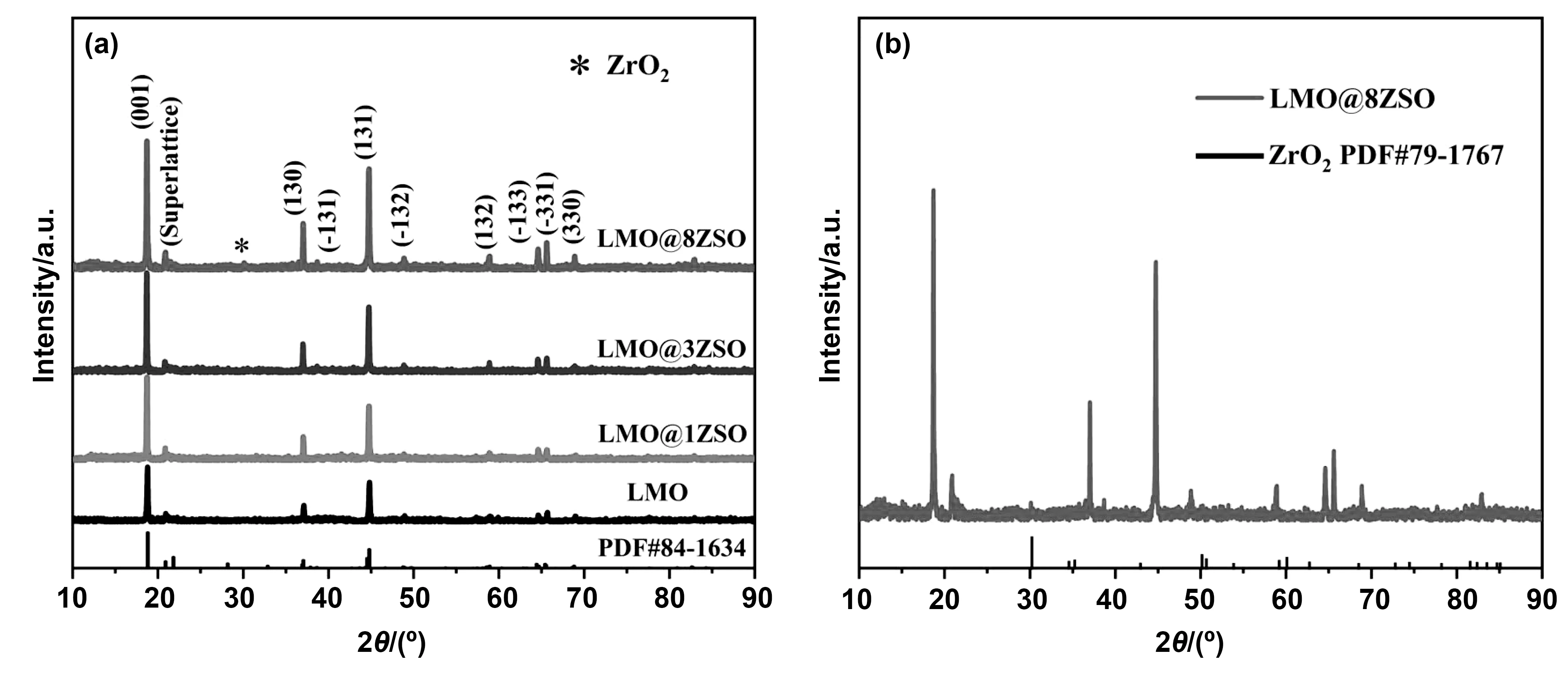
图2 (a)四种样品的X射线衍射图谱;(b)LMO@8ZSO的XRD衍射图谱与ZrO2标准PDF卡片对比图Fig.2 (a) X-ray diffraction patterns of four samples;(b) comparison of XRD diffraction pattern of LMO@8ZSO and ZrO2 standard PDF card
此外,在LMO@1ZSO和LMO@3ZSO的XRD图谱中,没有观测到包覆材料的衍射峰,推测可能是因为包覆量过于太少导致没有扫描出来。所以,加大了包覆量,制备了8%的ZSO对LMO进行包覆。在LMO@8ZSO的衍射图谱中,在2θ为30°左右观测到了一个比较弱的衍射峰。为了更好的观测,对LMO@8ZSO的XRD图谱进行放大处理,如图2(b)。由于所要合成的包覆材料氧离子导体Zr0.92Sr0.08O2没有匹配的标准PDF卡片,但其可以看做成在ZrO2中掺杂SrO,可以按ZrO2的PDF卡片进行匹配。经过PDF索引,发现与ZrO2标准PDF卡片PDF#79-1767的主峰位置基本吻合,为正方晶系,空间群为P42/nmc。此外,标准PDF卡片中ZrO2主峰的2θ为30.221°,而包覆材料的衍射峰角度为30.106°(图中带*的峰),说明包覆材料中ZrO2的衍射峰向小角度微微偏移,这是由于Sr的掺入导致的。由于Sr2+的离子半径(118pm)比Zr4+的离子半径(73pm)大,增大了晶面间距,从而使得衍射峰向左偏移。
2.1.2 SEM和EDS分析
如图3,(a)和(e)、(b)和(f)、(c)和(g)及(d)和(h)分别为纯LMO、LMO@1ZSO、LMO@3ZSO和LMO@8ZSO在500 nm和3 μm下进行的微观放大图。从图中可以看出,当包覆量较少时,LMO@1ZSO、LMO@3ZSO和纯LMO都可以看作为200nm左右的一次颗粒紧紧堆积起来的二次颗粒,且颗粒之间均存在空隙,不同之处在于包覆之后的材料,棱角分明,结晶度高,与上述XRD分析结果一致。当包覆量增加到8%时,存在部分比较大的颗粒,且在大颗粒的表面观察到了少量的细小颗粒。此外,为了更好地了解包覆之后材料中元素的分布,使用EDS能谱对LMO@8ZSO样品进行了元素测定。如图4,可以看出Mn、Zr和Sr元素均匀的分布在材料的表面,且没有出现成分的偏析和聚集现象。

图3 四种材料在不同放大倍数下的SEM图。其中,(a)和(e)为纯LMO;(b)和(f)为LMO@1ZSO;(e)和(g)为LMO@3ZSO;(d)和(h)为LMO@8ZSOFig.3 SEM images of four materials at different magnification:(a),(e) LMO;(b),(f) LMO@1ZSO;(e),(g) LMO@3ZSO;(d),(h) LMO@8ZSO

图4 LMO@8ZSO的EDS能谱图Fig.4 EDS maps of LMO@8ZSO
2.2 电化学性能分析
2.2.1 首次充放电曲线分析
图5(a)为LMO、LMO@1ZSO和LMO@3ZSO电极在2~4.8 V电压窗口,0.05C电流密度下测试的首次充放电曲线以及表1为3个电极的首次充放电比容量和库伦效率。可以看出,3个电极的充放电曲线基本保持一致。其中,纯LMO的首次充电比容量比LMO@1ZSO和LMO@3ZSO高,但首次库伦效率低于LMO@1ZSO和LMO@3ZSO,表明LMO@1ZSO和LMO@3ZSO两个电极,在首次充放电过程中发生的副反应较少,不可逆容量损失较小。

表1 纯LMO、LMO@1ZSO和LMO@3ZSO电极的首次充放电比容量和库伦效率Table 1 Initial charge-discharge capacity and coulomb efficiency of pure LMO,LMO@1ZSO and LMO@3ZSO electrodes

2.2.2 循环稳定性分析
如图6为纯LMO、LMO@1ZSO和LMO@3ZSO在2~4.8 V电压窗口,1C(250 mA/g)电流密度下测试的循环对比图。在1C电流密度下,纯LMO、LMO@1ZSO和LMO@3ZSO的首次放电比容量为121.59、136.88、139.31 mAh/g,其中LMO@1ZSO循环20次后放电比容量达到最大144.29 mAh/g,LMO@3ZSO循环13次后放电比容量达到最大143.74 mAh/g。循环100次后3个电极的放电比容量为53.92、113.24、106.17 mAh/g,对应的容量保持率为44.3%、82.7%、76.2%。从上面结果可以得出,纯LMO在循环过程中放电比容量一直持续下降,而LMO@1ZSO和LMO@3ZSO分别在前20次和前13次放电比容量一直处于上升的趋势,之后放电比容量才开始缓慢下降。所以,相比纯LMO,氧离子导体包覆改性的材料均表现出稳定的循环能力。这可以归因于在循环过程中,氧离子导体不断地抑制氧气的释放,使得氧不可逆损失降到最小,从而减小了容量衰减。
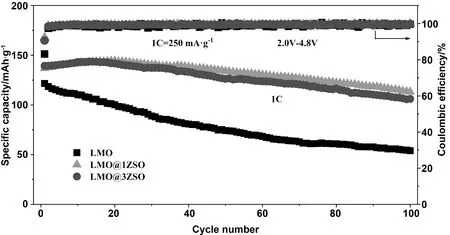
图6 纯LMO、LMO@1ZSO和LMO@3ZSO电极在1C电流密度下的循环性能图Fig.6 Cyclic performance of pure LMO,LMO@1ZSO and LMO@3ZSO electrodes at 1 C current density
放电平台电压衰减和容量衰减是Li2MnO3材料面临的最严重且最直观的问题,其可以从充放电曲线上直接看到。如图7,(a)、(c)和(e)分别为纯LMO、LMO@1ZSO和LMO@3ZSO在2~4.8 V电压范围,1C电流密度下,循环1次、20次、40次、60次、80次和100次的充放电曲线对比图。可以直观地看到,随着循环的进行,3个材料的充电平台逐渐上升,放电平台逐渐下降。在第一次充放电过程中,纯LMO的充电平台从3.6V左右开始,而LMO@1ZSO和LMO@3ZSO从3.3V左右开始,表明纯LMO电极的极化比改性材料的大,这不利于循环的后期进行。其次,纯LMO看不到放电平台,表明在循环第一次后,其结构都已经大部分塌陷,这归于纯LMO生成的是高度无序有缺陷的尖晶石相结构,它是没有电化学活性的,导致在随后的循环中放电平台电压和容量持续衰减[6]。当循环至20次时,很明显能看到LMO@1ZSO和LMO@3ZSO两个电极材料的放电电压平台变长,这是典型尖晶石相LiMn2O4的特征[21];其次放电比容量持续上涨,表明其在循环过程中的电化学可逆性强,从侧面也反映出经氧离子导体包覆后,LMO材料生成的尖晶石相是高度可逆的,可以理解为有序的无缺陷的尖晶石相结构;在循环100次后,两个电极材料的放电平台电压衰减都很小,表明经长期循环后其结构还能保持良好。这些结果可以归因于在循环过程中,(1)氧离子导体作为Li2MnO3的包覆层,阻碍了Mn2+与电解液的直接接触,防止Mn2+溶于电解液中;(2)氧离子导体将不可逆氧释放的氧离子返还给了主体材料,使得生成的尖晶石相是无缺陷的,具有电化学活性,从而防止了材料结构的后续坍塌,减小了放电平台电压衰减和容量衰减。
为了了解纯LMO和改性材料在循环过程中氧化还原反应的可逆性,对这3种电极材料的充放电曲线做了微分处理。如图7,(b)、(d)和(f)分别为3种电极材料在1C电流密度下,循环不同次数的dQ/dV曲线对比图。可以很明显观测到随着循环的进行,纯LMO的氧化还原峰逐渐的消失;而LMO@1ZSO和LMO@3ZSO两个电极在循环至20次之前,其氧化还原峰强度一直增加,表明生成的尖晶石相具有高度电化学活性,很容易发生氧化还原反应,这与其位于2.8 V的放电电压平台延长相对应;在循环100次后,也还能清楚地看到氧化还原峰,表明在循环过程中,相比纯LMO,LMO@1ZSO和LMO@3ZSO的材料结构保持得更好,这与上述放电电压平台衰减讨论的结果相一致。观测这几个图,可以发现,随着循环的进行,3个电极的氧化峰向着高电压区域移动,还原峰向着低电压区域移动,由此产生的电位差ΔV,可以反映电极在循环过程中发生氧化还原反应的可逆性[22]。纯LMO在第1次的电位差为1.535 V,而由于第100次的氧化还原峰消失,导致第100次的电位差不存在。反观改性材料,LMO@1ZSO和LMO@3ZSO在第1次的电位差分别为0.698和0.87 V,循环到100次时电位差分别为1.282和1.451 V,均比纯LMO第1次的电位差低。可见,经氧离子导体包覆后的材料的电极在循环过程中发生的氧化还原反应更加可逆。
2.2.3 循环伏安曲线分析
为了更加了解改性前后电极在循环过程中的氧化还原反应,对纯LMO和LMO@1ZSO分别进行了循环伏安(CV)测试。如图8(a)和(b)分别为纯LMO和LMO@1ZSO在2~4.8 V的电压窗口下,扫描速度为0.1 mV/s,测试前3圈的CV曲线。总体来看,两个电极材料的CV曲线基本保持一致。在首次循环中,位于4.7 V的氧化峰代表着电极中Li2O的去除,在还原过程中没有找到与之对应的还原峰,表明这一氧化反应不可逆;且LMO@1ZSO这一氧化峰强度更低,代表着其O2-→O2的反应强度减少,这与上述首次充放电曲线和微分电容曲线分析结果一致。其次,位于3.4和2.8 V左右的还原峰对应于层状相和尖晶石相,且在第二圈看到了与之对应的氧化峰,分别位于4.4和3.1 V左右,表明这两对氧化还原反应是可逆的,对应层状Mn3+↔Mn4+和尖晶石状Mn3+↔Mn4+[23]。仔细观察,改性后的电极在2.8 V的还原峰强度较尖锐,表明其生成的尖晶石相结构更容易进行电化学反应,不同于纯LMO生成的的缺陷无活性尖晶石相。循环过程中峰的偏移程度及曲线的重叠度可以反映出电极氧化还原反应的可逆性程度[24]。其中,位于2.8 V左右的还原峰,纯LMO向着更低的电位偏移,而LMO@1ZSO的还原峰几乎没有移动;而且LMO@1ZSO的CV曲线重叠度比纯LMO的要更好。表明对比纯样,改性材料的电极发生氧化还原反应的过程更加可逆,这与上述不同循环次数下的dQ/dV曲线讨论结果相一致。

图8 (a)和(b)分别为纯LMO和LMO@1ZSO电极的循环伏安曲线图Fig.8 The CV curves of (a) pure LMO and (b) LMO@1ZSO electrodes
3 结 论
本文先采用溶胶凝胶法制备出了Li2MnO3正极材料,然后采用湿化学法将Zr0.92Sr0.08O2溶液包覆在Li2MnO3材料表面,制备出了LMO@ZSO多组分复合材料,经过XRD、SEM和EDS测试表明Zr0.92Sr0.08O2包覆层被制备出来且成功引入到了Li2MnO3材料表面,且所有材料都保持着良好的层状结构。通过电化学测试分析,氧离子导体Zr0.92Sr0.08O2包覆后的正极材料表现出更出色的电化学性能。在2.0~4.8 V电压窗口,0.05C电流密度下,LMO@1ZSO正极材料的首次充放电效率为65.86%,而纯LMO只有62.18%;在1C电流密度下循环100次,LMO@1ZSO正极材料的容量保持率为82.7%,而纯LMO只有44.3%。由于氧离子导体具有传输氧离子的特性,在循环过程中,它会使一部分的氧离子重新返回体内,缓解了氧的不可逆释放,提高了首次充放电效率,且生成的尖晶石相具有电化学活性,可以很容易发生氧化还原反应,防止了后续结构的不断坍塌,缓解了放电平台电压和容量衰减,从而表现出更好的循环稳定性。
