3种类型秸秆降解对黑土微生物群落及功能的影响
2023-10-17郭悦万雨欣徐伟慧于志丹王志刚
郭悦,万雨欣,徐伟慧,于志丹,王志刚
3种类型秸秆降解对黑土微生物群落及功能的影响
郭悦1,2,万雨欣1,2,徐伟慧1,2,于志丹1,王志刚1,2
(齐齐哈尔大学 1. 生命科学与农林学院,2. 黑龙江省农业微生物制剂产业化技术创新中心,黑龙江 齐齐哈尔 161006)
秸秆的降解主要依赖于微生物对木质纤维素残体的作用.黑土土质肥沃,富含微生物,利用宏基因组测序了解秸秆降解过程中黑土微生物的变化具有重要意义.以3种东北黑土区典型农作物秸秆即大豆、水稻、玉米秸秆为材料,加入到黑土中,在30℃条件下进行微宇宙实验,分析在秸秆降解过程中土壤微生物群落和功能的变化.结果表明,大豆、水稻和玉米秸秆在黑土环境中富集了不同的微生物.大豆秸秆主要富集放线菌(Actinobacteria)、绿弯菌(Chloroflexi)、COG1858,水稻秸秆主要富集拟杆菌(Bacteroidetes)、疣微菌门(Verrucomicrobia)、COG1472、COG3250,玉米秸秆主要富集变形菌(Proteobacteria)、酸杆菌(Acidobacteria)、ENOG410XNTA.不同类型的秸秆降解过程中会富集不同的木质纤维素降解菌,木质纤维素降解相关的功能途径和酶的丰度也有所增加.
黑土微生物;秸秆降解;宏基因组测序;微生物群落和功能
黑土具有良好的土壤性状,富含有机质,土壤肥沃,肥力可达普通土壤的10倍[1],适合农作物生长[2].黑土地区是中国主要的粮食产区,非常适合大豆、玉米和水稻的种植.此外,黑土中有丰富的微生物种类,是秸秆降解的主要驱动因素[3-4].存在大量降解秸秆纤维素的微生物,在农业发展和土壤生态学中发挥着重要作用[5].木质纤维素是植物残体的重要组成部分,是秸秆的主要成分,其丰富的碳源对土壤碳循环至关重要[6-7].秸秆中的碳可以为作物生长提供有利的土壤条件,也可以提高土壤肥力和作物产量[8-9],对秸秆进行生物降解可以提高秸秆的利用率.秸秆的降解过程需要许多酶的参与,具有产酶能力的菌富集在秸秆表面和内部并在作物秸秆中起到了分解者的作用[10].大量研究报道了木质纤维素降解菌,它们产生纤维素降解酶,能够将大的纤维素聚合物降解为小分子化合物或单体化合物,最终转化为CO2和H2O[11].细菌具有抗逆性强、繁殖快等特点[12],因此在木质纤维素降解中备受青睐.芽孢杆菌属()作为研究中最常见的菌属在木质纤维素降解中发挥了重要作用[13],其他一些菌属同样具有产木质纤维素降解相关酶的能力,如普氏菌属()、栖瘤胃解纤维素菌属()和梭状菌属()[14-15]、节杆菌属()[16]等.秸秆较高的碳氮比环境会限制微生物的繁殖[17],但放线菌的固氮能力可能在微生物驱动的植物残体分解过程中提高氮的有效性[18].放线菌作为重要的细菌门是土壤细菌中分布最广的门之一,对植物残体的降解能力也是众所周知的[19-21],放线菌具有更好的适应能力,在秸秆木质纤维素降解研究中具有更重要的生理生态作用[22].
大豆、水稻和玉米是东北黑土区的主要粮食作物.在一年一度的农作物收获之后,剩余的农作物秸秆造成了一些环境问题,如秸秆燃烧会释放有害气体.因此,如何通过无污染的方式处理秸秆资源是一个亟待解决的问题.木质纤维素降解是非常复杂的过程,需要微生物产生的各种酶共同发挥作用[23].本文结合东北黑土区的实际情况,将大豆、水稻和玉米秸秆添加到黑土中进行微宇宙实验.在微环境驯化120 d的条件下,分析了大豆、水稻和玉米秸秆降解对黑土微生物多样性、群落结构和功能的影响,旨在为秸秆的无害化处理提供理论依据.
1 材料与方法
1.1 实验材料
黑土采集于黑龙江省齐齐哈尔市克山县(48°21′43″N,126°03′05″E).从地面(0~20 cm表土)取样并充分混合以形成复合样品.随后,将样品在室温下风干,并通过3 mm筛,在处理之前除去植物碎片和大石头.土壤理化性质见表1.

表1 黑土的理化性质
注:数据表示为平均值±标准偏差.下同.
大豆、水稻、玉米秸秆采集于黑龙江省齐齐哈尔市建华区曙光村(47°35′99″N,123°92′46″E).将大豆、水稻、玉米秸秆切成3~4 cm的小段,并在80℃的烘箱中烘干至恒质量.秸秆理化性质见表2.

表2 秸秆的理化性质 (%)
注:不同小写字母表示处理之间有显著差异<0.05.下同.
1.2 样品采集及处理
将大豆、水稻、玉米秸秆分别与黑土以10∶1的比例在盆中均匀混合,分别设为DH(大豆秸秆与黑土混合)、SH(水稻秸秆与黑土混合)、YH(玉米秸秆与黑土混合)组,并置于30℃的培养箱中进行微宇宙实验.整个过程持续120 d,水分保持在60%左右.分别在第0天和第120天收集秸秆样品表面的土壤,用于宏基因组测序,每个处理设3个重复,同时设置不加秸秆的黑土作为对照组(CK).
1.3 宏基因组学测序与统计分析
使用E.Z.N.A.®Soil DNA Kit试剂盒提取土壤微生物总DNA.宏基因组测序工作由上海美吉生物医药科技有限公司使用Illumina NovaSeq测序平台完成[24].具体流程为:(1)文库分子一端与引物碱基互补,经过一轮扩增,将模板信息固定在芯片上;(2)固定在芯片上的分子另一端随机与附近的另外一个引物互补,也被固定住,形成“桥”;(3)PCR扩增,产生DNA簇;(4)DNA扩增子线性化成为单链;(5)加入改造过的DNA聚合酶和带有4种荧光标记的dNTP,每次循环只合成1个碱基;(6)用激光扫描反应板表面,读取每条模板序列第1轮反应所聚合上去的核苷酸种类;(7)将“荧光基团”和“终止基团”化学切割,恢复3′端粘性,继续聚合第2个核苷酸;(8)统计每轮收集到的荧光信号结果,获知模板DNA片段的序列.
来自土壤的DNA样品在Illumina NovaSeq 6000平台上进行测序,每个样品平均6 Gb.(1)所有的原始数据都通过Fastp(https://github.com/OpenGene/fastp,version 0.20.0)对reads 3′端和5′端的adapter序列进行质量剪切,去除剪切后长度小于50 bp、平均碱基质量值低于20以及含N碱基的reads,保留高质量的pair-end reads和single-end reads;(2)过滤后的读数用MEGAHIT(https://github.com/voutcn/megahit,version 1.1.2)对优化序列进行拼接组装,在拼接结果中筛选≥300 bp的contigs作为最终的组装结果;(3)用Prodigal(https://github.com/hyattpd/Prodigal)来预测开放阅读框架(ORF),选择核酸长度≥100 bp的基因,并将其翻译为氨基酸序列;(4)使用CD-HIT(http://weizhongli-lab.org/cd-hit/,version 4.6.1)对所有样品预测出来的基因序列进行聚类,去除冗余,并获得唯一的基因目录;(5)使用SOAPaligner(https://github.com/ShujiaHuang/SOAPaligner)通过比较干净的读数和基因目录来计算基因深度和相对丰度.分别将每个样品的高质量reads与非冗余基因集进行比对(95% identity),统计基因在对应样品中的丰度信息;(6)使用Diamond(http://www.diamondsearch.org/index.php,version 0.8.35)将非冗余基因集的氨基酸序列与NR数据库进行比对(BLASTP比对参数设置期望值e-value为1e-5),并通过NR库对应的分类学信息数据库获得物种注释,然后使用物种对应的基因丰度总和计算该物种的丰度;(7)使用Diamond(http://www.diamondsearch.org/index.php,version 0.8.35)将非冗余基因集的氨基酸序列与EggNOG(evolutionary genealogy of genes:Non-supervised Orthologous Groups)数据库进行比对(BLASTP比对参数设置期望值e-value为1e-5),获得基因对应的COG(Clusters of orthologous groups of proteins,直系同源蛋白簇)功能,然后使用COG对应的基因丰度总和计算该COG的丰度.原始数据上传至NCBI网站SRA数据库(Accession Number:PRJNA918990).
所有数据均在Excel 2019整理,通过SPSS 22.0(IBM,Armonk,New York,USA)进行所有统计学分析.数据分析采用单因素方差分析(ANOVA),<0.05表示具有统计学意义.在属水平上计算微生物的多样性Chao、Shannon指数;使用基于bray-curtis距离算法的PCoA分析(主坐标分析)检验样本间微生物群落结构的相似性.基于基因的物种分类学注释,比对NR数据库获得样本物种的分类学注释信息,使用Reads Number计算丰度.在EggNOG数据库中获得基因对应的COG,然后使用COG对应的基因丰度总和计算该COG的丰度.使用GraphPad Prism8.0.2软件绘制柱形图,使用Origin 2021软件绘制柱状堆叠图,使用TBtools软件(Toolbox for Biologists v1.09876)绘制丰度热图.
2 结果与分析
2.1 3种类型秸秆降解对黑土微生物群落多样性的影响
第0天和第120天土壤微生物的多样性指数见图1.由图1可见,随着时间的推移,土壤微生物的多样性发生了显著变化.在秸秆降解第0天取样的3个处理组(DH,SH,YH)和CK组的多样性在整个过程中是最低的.秸秆降解后,黑土中微生物群落的多样性显著增加,没有添加秸秆的CK组变化较小.DH,SH,YH组的Chao指数在第120天分别比第0天升高了371.33,365.00,333.67(见图1a),Shannon指数分别升高了0.50,0.38,0.52(见图1b).这些结果表明,秸秆降解会增加黑土微生物的多样性.
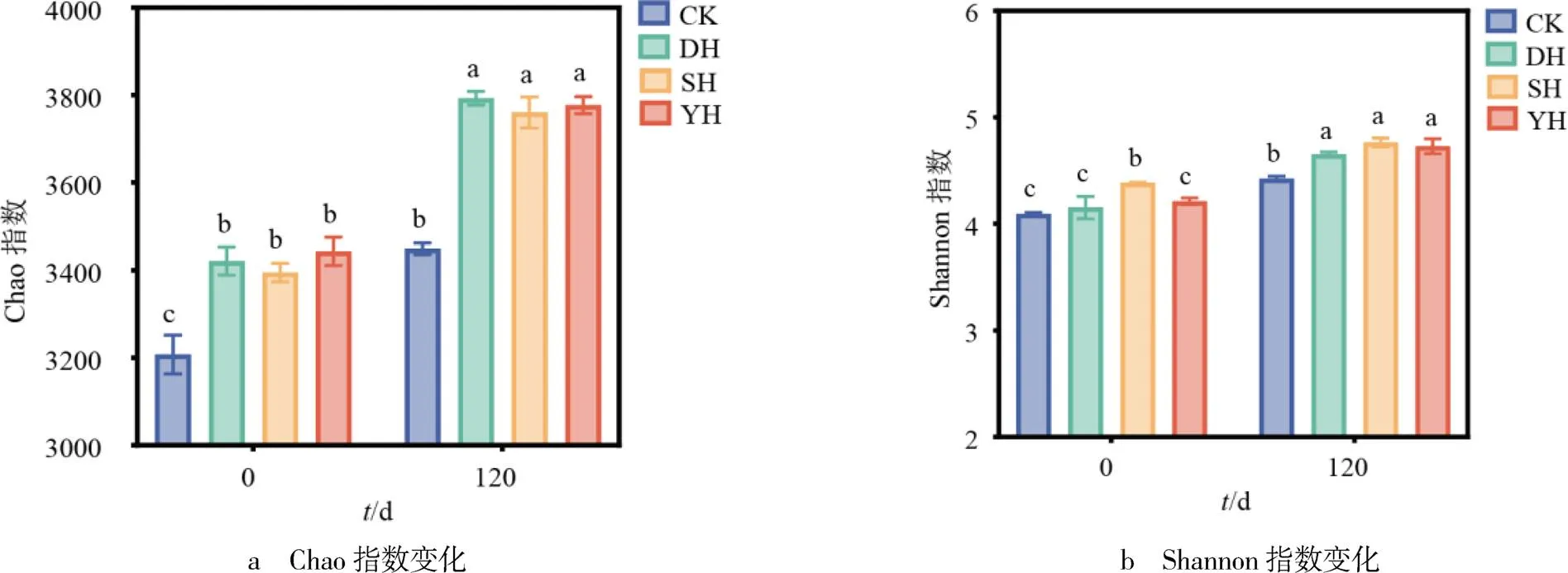
图1 3种类型秸秆降解过程中黑土微生物群落α多样性的变化
基于bray-curtis距离算法的PCoA分析不同处理间微生物群落结构的多样性结果见图2.由图2a可见,第0天的DH,SH,YH组与CK组并未完全分离开,表示在该阶段各个处理组之间差异较小,并不显著.由图2b可见,第120天的DH,SH,YH组与CK组明显分离,说明在第120天DH,SH,YH组与CK组彼此之间均存在显著差异.
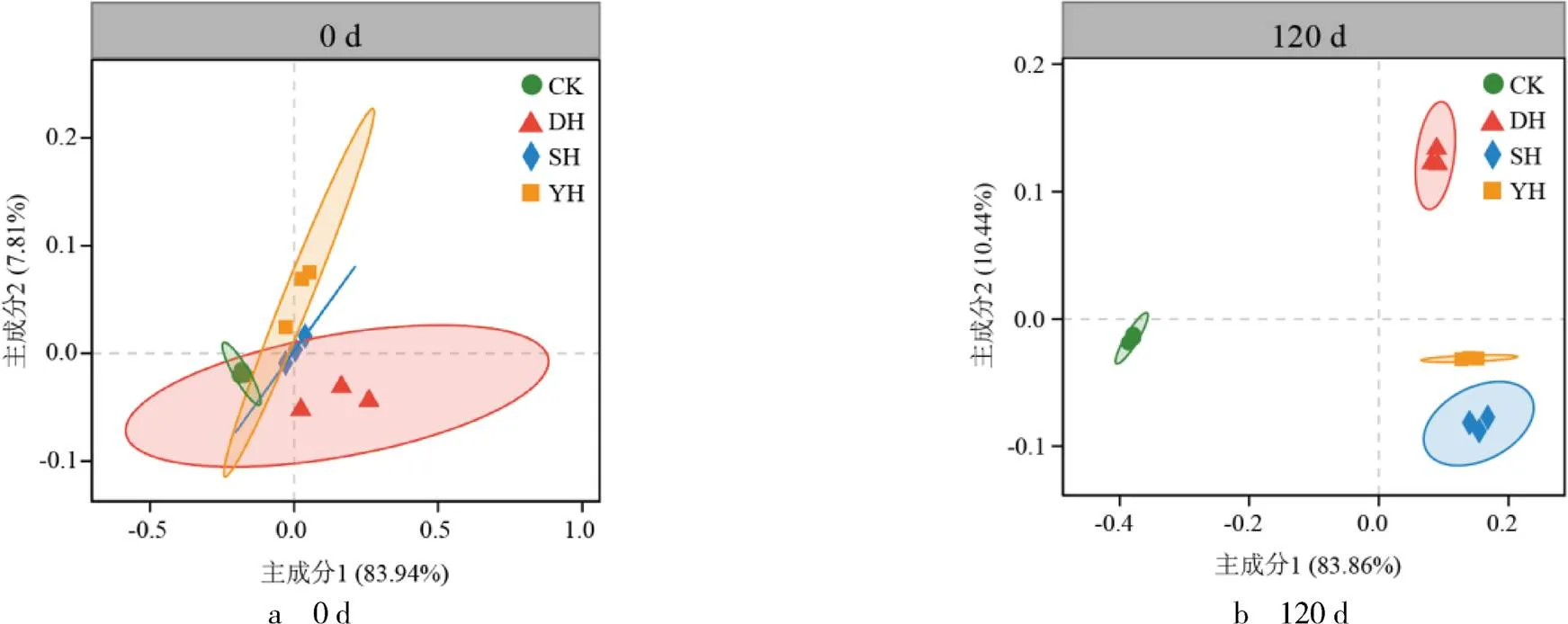
图2 3种类型秸秆降解过程中黑土微生物群落β多样性的变化
注:图中椭圆为置信椭圆.
2.2 3种类型秸秆降解对黑土微生物群落组成的影响
通过NR物种注释在门水平上对微生物群落结构进行分析(见图3),将相对丰度小于0.01的菌门合并为others后,除未分类的细菌,共发现了11个相对丰度较高的菌门.其中放线菌门(Actinobacteriota)、变形菌门(Proteobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)、厚壁菌门(Firmicutes)为优势菌门.在120 d的实验周期内,CK组的各个菌门的丰度变化较小.放线菌门作为主要菌门之一,在DH,SH,YH组都显著减少,分别减少了35.07%,44.12%,44.68%.同为优势菌的变形菌门,在第120天时,DH,SH,YH组分别比CK高出15.62%,16.60%,21.92%.由此可见,碳氮比最高的玉米秸秆富集了更多的变形菌.3种秸秆中碳氮比最小的大豆秸秆与另外2组相比可以富集更多的绿弯菌门,在第120天的相对丰度为21.65%.
对属水平上前30名的菌属变化进行分析(见图4).由图4可见,DH,SH,YH组均显著富集了固氮菌属()、厌氧菌属()以及一些属水平未分类的变形菌(Proteobacteria)、绿弯菌(Chloroflexi)和酸杆菌(Acidobacteria)等,这些菌的丰度在第120天与第0天相比均有显著增加.而芽孢杆菌属()、泛菌属()、假诺卡氏菌()等菌株的丰度显著减少.从不同秸秆处理组的差异来看,鞘氨醇单胞菌属()在DH和YH组中显著富集,尤其是在YH组,而在SH组的丰度显著减少;假单胞菌()在DH和SH组显著减少,而在YH组则差异较小.
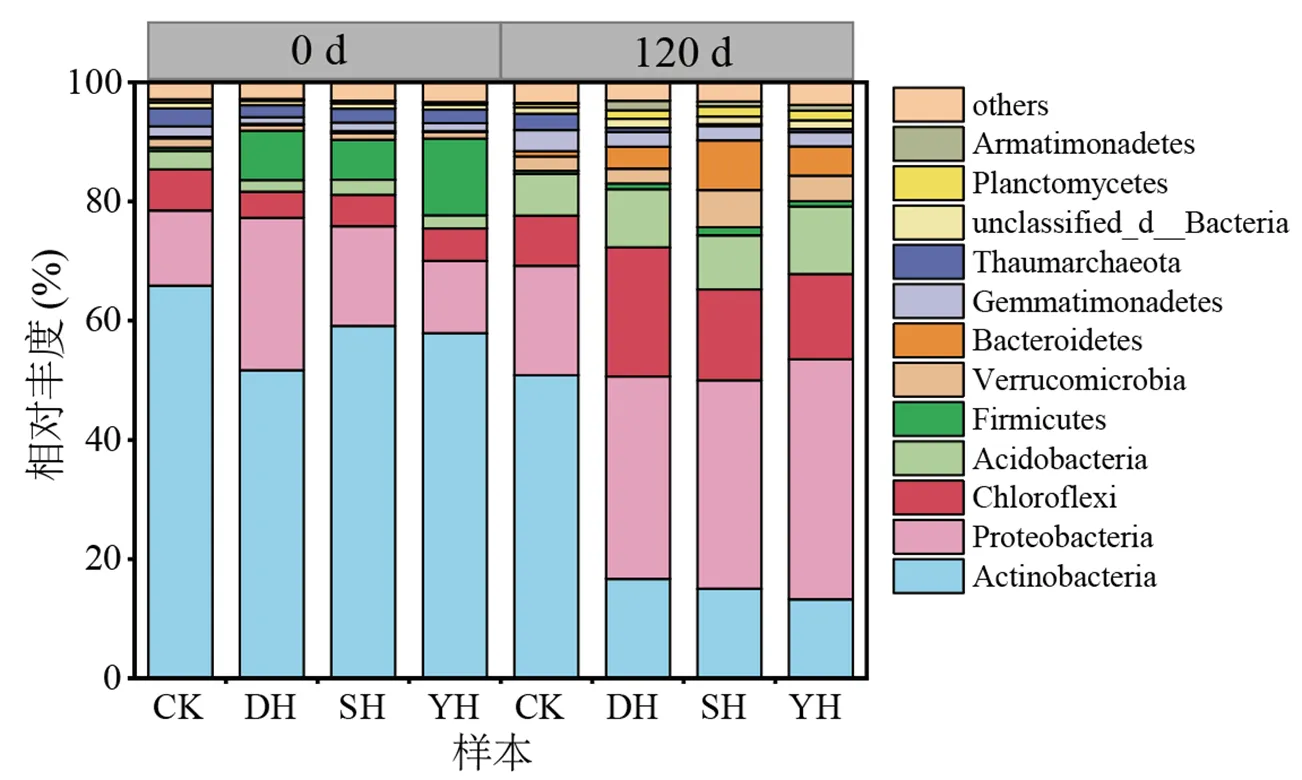
图3 3种秸秆降解过程中黑土微生物门水平群落结构的变化
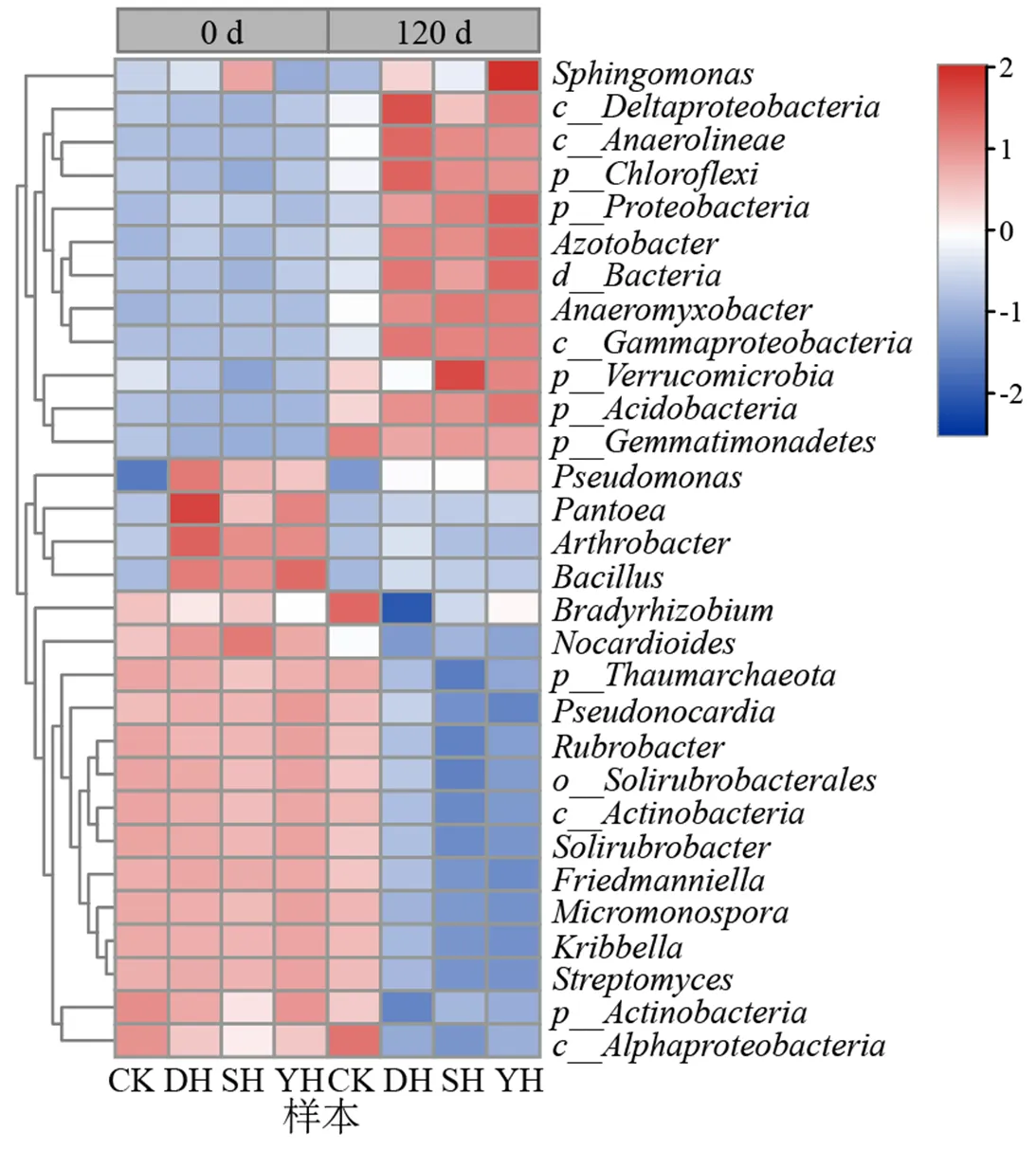
图4 3种秸秆降解过程中黑土微生物属水平群落丰度热图
2.3 3种类型秸秆降解对黑土微生物群落功能的影响
比对Egg NOG数据库后,对DH,SH,YH处理组以及CK组的细菌功能进行了COG功能注释,其中19个COG功能属于新陈代谢,分别包括碳水化合物的运输和代谢、能源生产和转化以及无机离子运输和代谢.绘制丰度前20功能的热图(见图5),与水解酶家族相关的COG1472,COG3250,COG3693在DH,SH,YH组均有所增加,其中在SH组中增加最多,分别是第0天的2.12,3.92,12.77倍.与糖苷水解酶家族相关的ENOG410XNTA和COG2730在3组中也显著增加,其中SH和YH富集到更多,达到了第0天的8倍以上,但同样属于糖苷水解酶家族的COG1486在DH,SH,YH组中均有所减小,其中YH最少,仅为第0天的0.61倍.与-半乳糖苷酶(-galactosidase)相关的COG1874在SH组中显著富集,是第0天的2.72倍.与其他处理组相比,DH显著富集了与细胞色素C过氧化物酶相关的COG1858;YH显著富集了与具有过氧化氢酶和广谱过氧化物酶活性的双功能酶相关的COG0376.
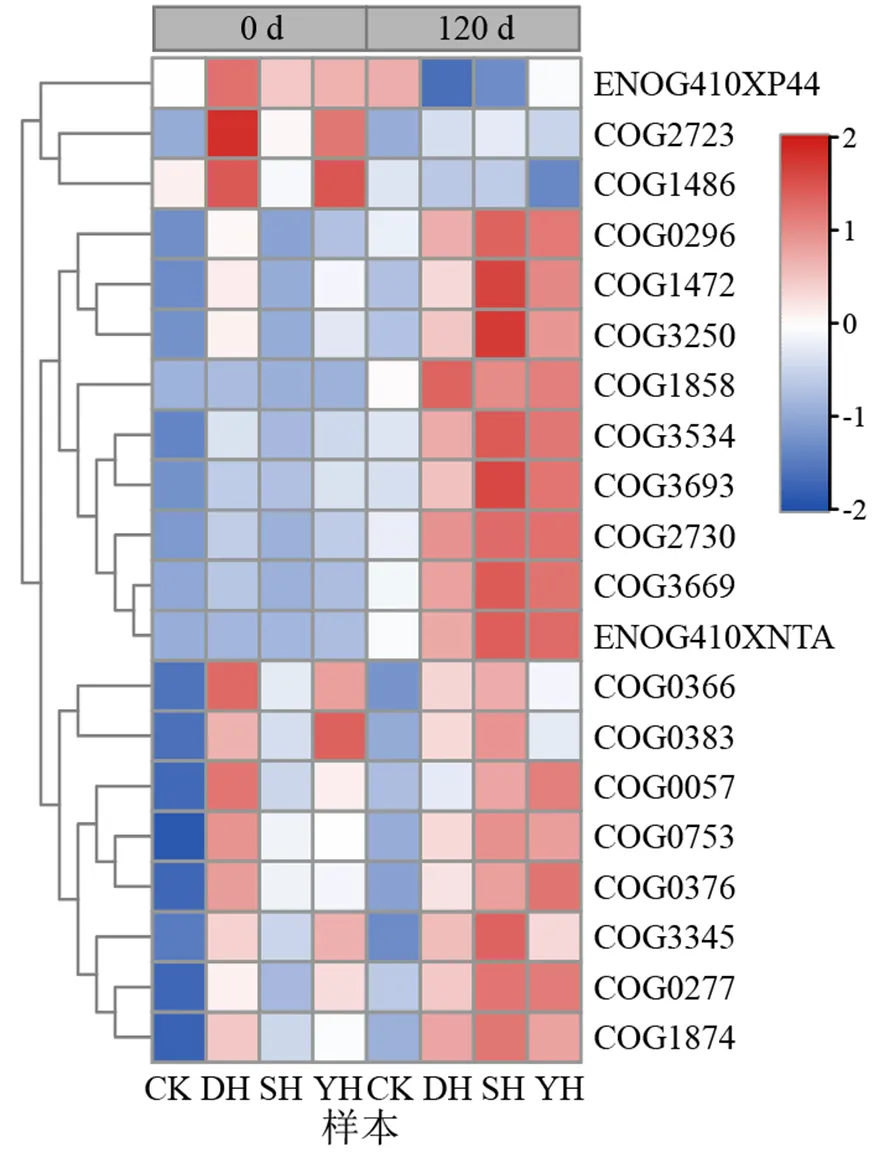
图5 3种秸秆降解过程中COG功能注释丰度热图
3 讨论与结论
土壤微生物在有机物分解中起到重要作用[25].农作物秸秆作为一种优质的纤维素结构体,可以为土壤碳循环提供优质碳源.同时,土壤中丰富的微生物群落共同利用秸秆中的木质纤维素进行生命活动.3种类型秸秆降解会增加土壤细菌的多样性,显著改变黑土中微生物的群落结构[26-27].在秸秆降解过程中,变形菌门长期作为土壤中优势菌门[26,28-30].同时,秸秆降解会降低土壤中放线菌门的相对丰度[30-31].拟杆菌门作为另一个优势菌门,是分解半纤维素和木聚糖的重要菌门[32].因此,DH,SH,YH组中,拟杆菌门的丰度都显著大于CK,其中以水稻秸秆降解过程中DH组丰度最高.在属水平上,固氮菌属()显著增加,可以将空气中植物无法吸收的氮气转化成可以吸收利用的氨或者其他含氮有机物[33],与第0天比较,第120天固氮菌显著增加,固氮微生物可以提供氮素,缓解因秸秆过高的碳氮比而不利于降解的问题[4,34].同时,固氮菌属在YH中更加显著富集,这是玉米秸秆含有更高的碳氮比导致的.链霉菌属()作为放线菌门的重要菌属,在秸秆降解过程中具有重要的生态作用[35],并且在植物残体降解过程中保持稳定存在.克雷伯氏菌()是肠杆菌科常见的菌属,在木质纤维素降解过程中同样发挥了重要作用[36].秸秆的加入引起了土壤微生物群落结构和多样性的变化,这些变化是由于秸秆的加入增加了土壤的碳含量,改变了土壤的理化性质,导致了土壤环境的巨大变化.不同类型的秸秆含有不同的碳含量,因此微生物的富集及其对土壤理化性质的影响存在差异[37].通过COG注释对功能进行分析发现,注释到的前20个COG中有19个属于新陈代谢,其中15个属于碳水化合物的运输和代谢,说明秸秆降解过程中需要大量的碳水化合物的相关酶,如-淀粉酶、-半乳糖苷酶、-葡萄糖苷酶、过氧化氢酶和糖苷水解酶等[38],该过程需要多种酶进行协同作用达到将其资源化利用的效果.
综上所述,通过宏基因组学测序结果表明,不同类型的秸秆降解过程中会富集不同的木质纤维素降解菌,木质纤维素降解相关的功能途径和酶的丰度也有所增加.本文为今后黑土地区的秸秆降解研究提供了理论依据,对减少环境污染具有重要意义.
[1] 林琪.让黑土地更加“肥美”[J].环境,2021(8):75-78.
[2] 崔宁波,生世玉.粮食安全视角下的东北黑土区耕地生态补偿研究[J].浙江农业学报,2021,33(9):1759-1769.
[3] Chidthaisong A,Conrad R.Pattern of non-methanogenic and methanogenic degradation of cellulose in anoxic rice field soil[J]. FEMS Microbiology Ecology,2000,31(1):87-94.
[4] Bao Y,Feng Y,Stegen J C,et al.Straw chemistry links the assembly of bacterial communities to decomposition in paddy soils[J].Soil Biology and Biochemistry,2020,148:107866.
[5] Enjun K,Minggang X,Colinet G,et al.Degradation characteristics of maize straw under different buried depths in northeast black soil and their effects on soil carbon and nitrogen[J].International Journal of Agriculture Biology,2020,24:77-84.
[6] Chen S,Xia Y,Zhang B,et al.Disassembly of lignocellulose into cellulose,hemicellulose,and lignin for preparation of porous carbon materials with enhanced performances[J].Journal of Hazardous Materials,2021,408:124956.
[7] Wang X,Yang Y,Zhang X,et al.To make biofuel:cutting the lignin or loosening lignin′s grip?[J].Scientia Agricultura Sinica,2015,48(2):229-240.
[8] Li Y,Feng H,Dong Q,et al.Ammoniated straw incorporation increases wheat yield,yield stability,soil organic carbon and soil total nitrogen content[J].Field Crops Research,2022,284:108558.
[9] Wang H,Shen M,Hui D,et al.Straw incorporation influences soil organic carbon sequestration,greenhouse gas emission,and crop yields in a Chinese rice(L.)-wheat(L.)cropping system[J].Soil and Tillage Research,2019,195:104377.
[10] Wickings K,Grandy A S,Reed S C,et al.The origin of litter chemical complexity during decomposition[J].Ecology Letters,2012,15(10):1180-1188.
[11] Ma X,Gao M,Li Y,et al.Production of cellulase bythrough fermentation of spent mushroom substance:glucose inhibition and elimination approaches[J].Process Biochemistry,2022,122:26-35.
[12] Hasunuma T,Okazaki F,Okai N,et al.A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology[J].Bioresource Technology,2013,135:513-522.
[13] 江高飞,暴彦灼,杨天杰,等.高温秸秆降解菌的筛选及其纤维素酶活性研究[J].农业环境科学学报,2020,39(10):2465-2472.
[14] Pan Y,Zheng X,Xiang Y.Structure-function elucidation of a microbial consortium in degrading rice straw and producing acetic and butyric acids via metagenome combining 16S rDNA sequencing[J].Bioresource Technology,2021,340:125709.
[15] 张蕴琦,徐凤花,张书敏,等.水稻秸秆降解菌系的筛选及其菌群组成分析[J].江苏农业科学,2017,45(8):257-260.
[16] 万文结,刘月,薛芷筠,等.纤维素降解菌HW-17的纤维素降解特性及纤维素酶学性质[J].环境科学学报,2017,37(10):3679-3686.
[17] Seneviratne G.Litter quality and nitrogen release in tropical agriculture:a synthesis[J].Biology and Fertility of Soils,2000,31(1):60-64.
[18] Subramaniam G,Arumugam S,Rajendran V.Plant growth promoting Actinobacteria:a new avenue for enhancing the productivity and soil fertility of grain legumes[M].Singapore:Springer Singapore,2016:123-145.
[19] Bao Y,Dolfing J,Wang B,et al.Bacterial communities involved directly or indirectly in the anaerobic degradation of cellulose[J].Biology and Fertility of Soils,2019,55(3):201-211.
[20] Abdulla H M,El-Shatoury S A.Actinomycetes in rice straw decomposition[J].Waste Management,2007,27(6):850-853.
[21] Lewin G R,Carlos C,Chevrette M G,et al.Evolution and ecology of actinobacteria and their bioenergy applications[J].Annual Review of Microbiology,2016,70(1):235-254.
[22] Bao Y,Dolfing J,Guo Z,et al.Important ecophysiological roles of non-dominant actinobacteria in plant residue decomposition,especially in less fertile soils[J].Microbiome,2021,9(1):84.
[23] Gahfif O,Souagui Y,Azzouz Z,et al.Isolation and screening of fungal culture isolated from algerian soil for the production of cellulase and xylanase[J].Journal of Drug Delivery Therapeutics,2020,10:108-113.
[24] Du K,Yang F,Zhang J T,et al.Comparative genomic analysis of five freshwater cyanophages and reference-guided metagenomic data mining[J].Microbiome,2022,10(1):128.
[25] McGuire K L,Treseder K K.Microbial communities and their relevance for ecosystem models:decomposition as a case study[J].Soil Biology and Biochemistry,2010,42(4):529-535.
[26] Bu R,Ren T,Lei M,et al.Tillage and straw-returning practices effect on soil dissolved organic matter,aggregate fraction and bacteria community under rice-rice-rapeseed rotation system[J].Agriculture,Ecosystems and Environment,2020,287:106681.
[27] Zhao S,Li K,Zhou W,et al.Changes in soil microbial community,enzyme activities and organic matter fractions under long-term straw return in north-central China[J].Agriculture,Ecosystems and Environment,2016,216:82-88.
[28] Bai N,Zhang H,Zhou S,et al.Long-term effects of straw return and straw-derived biochar amendment on bacterial communities in soil aggregates[J].Scientific Reports,2020,10(1):7891.
[29] Song Y,Li X,Xu M,et al.Does biochar induce similar successions of microbial community structures among different soils?[J].Bulletin of Environmental Contamination and Toxicology,2019,103(4):642-650.
[30] Su Y,Lv J L,Yu M,et al.Long-term decomposed straw return positively affects the soil microbial community[J].Journal of Applied Microbiology,2020,128(1):138-150.
[31] Sun D,Meng J,Xu E G,et al.Microbial community structure and predicted bacterial metabolic functions in biochar pellets aged in soil after 34 months[J].Applied Soil Ecology,2016,100:135-143.
[32] Maarastawi S A,Frindte K,Linnartz M,et al.Crop rotation and straw application impact microbial communities in Italian and Philippine soils and the rhizosphere of[J].Frontiers in Microbiology,2018,9:1295.
[33] 周倩,黄安诚.植物根系化合物调控微生物菌群研究进展[J].植物生理学报,2020,56(11):2288-2295.
[34] Bao Y,Guo Z,Chen R,et al.Functional community composition has less environmental variability than taxonomic composition in straw-degrading bacteria[J].Biology and Fertility of Soils,2020,56(6):869-874.
[35] Anderson I,Abt B,Lykidis A,et al.Genomics of aerobic cellulose utilization systems in actinobacteria[J].PLoS One,2012,7(6):e39331.
[36] Jiang J,Tun H M,Mauroo N F,et al.Complete genome sequence and comparative genome analysis of Klebsiella oxytoca HKOPL1 isolated from giant panda feces[J].BMC Research Notes,2014,7:827.
[37] Jiang X,Denef K,Stewart C E,et al.Controls and dynamics of biochar decomposition and soil microbial abundance,composition,and carbon use efficiency during long-term biochar-amended soil incubations[J].Biology and Fertility of Soils,2015,52:1-14.
[38] 姚兵莉,张继宁,蓝娜娜,等.黑曲霉在生物质废弃物资源化中的应用[J].生物加工过程,2019,17(4):392-401.
Effects of degradation of three types of straw on microbial communities and functions in black soil
GUO Yue1,2,WAN Yuxin1,2,XU Weihui1,2,YU Zhidan1,WANG Zhigang1,2
(1. School of Life Sciences,Agriculture and Forestry,2. Heilongjiang Provincial Technology Innovation Center of Agromicrobial Preparation Industrialization,Qiqihar University,Qiqihar 161006,China)
Straw degradation depends mainly on bacterial and fungal actions on lignocellulosic residues.Black soil is fertile and rich in microorganisms.Therefore,it is of great significance to understand the changes in soil microorganisms during straw degradation by metagenomic sequencing.Three typical crop straw,namely soybean,rice and corn straws,from the black soil region of northeast China were added to black soil at 30℃ for microcosm experiments to analyze the changes in soil microbial communities and functions during the straw degradation process.The results showed that soybean,rice and corn straw were enriched with different microorganisms in black soil.Soybean straw was mainly enriched with Actinobacteria,Chloroflexi and COG1858.Rice straw was mainly enriched with Bacteroidetes,Verrucomicrobia,COG1472 and COG3250.Corn straw was mainly enriched with Proteobacteria,Acidobacteria,ENOG410XNTA.The processes of different straw degradation were enriched with different lignocellulose-degrading bacteria,and the abundance of functional pathways and enzymes related to lignocellulose degradation increased.
black soil microorganisms;straw degradation;metagenomic sequencing;microbial communities and functions
1007-9831(2023)09-0051-08
Q93
A
10.3969/j.issn.1007-9831.2023.09.012
2023-06-29
国家自然科学基金项目(31870493);黑龙江省重点研究开发项目(GA21B007);齐齐哈尔大学创新创业训练计划项目(201810232167)
郭悦(1997-),女,黑龙江齐齐哈尔人,在读硕士研究生,从事环境微生物学研究.E-mail:1275598797@qq.com
王志刚(1980-),男,内蒙古赤峰人,教授,博士,从事微生物资源与开发研究.E-mail:wangzhigang@qqhru.edu.cn
