CRISPR/Cas9技术在工业微生物中的应用*
2023-07-19张才达祁永浩米雅萱张蕴之秦浩杰刘东李小兵任丽梅
张才达 祁永浩 米雅萱 张蕴之 秦浩杰 刘东 李小兵 任丽梅**
(1)石家庄学院化工学院,河北省高校微生物制药应用技术研发中心,石家庄 050035;2)美邦美和生物科技有限公司,石家庄 050000)
基因编辑作为代谢工程、医学、农学等领域的重要技术,一直以来都是研究的热点。传统的基因编辑方法如同源重组,存在打靶效率低、操作烦琐等问题。随后出现的锌指核酶技术(zinc-finger nucleases,ZFN)[1]和转录激活因子效应蛋白核酸酶技术(transcription activator-like effector nucleases,TALEN)[2],虽一定程度上提高了编辑效率,简化了编辑步骤,然而这两个技术也因难度较大、费用较高等原因,应用范围受到限制。相对而言,近年来发展的CRISPR技术,具有操作难度小、编辑成本低、打靶效率高、无痕编辑等优点,特别适用于微生物底盘细胞的编辑。改造后的或新创造的底盘细胞,更有利于实现特定的生物功能从而实现目标产物的大量积累。本文主要简述以规律间隔成簇短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)/Cas9为基础而衍伸出的各种基因编辑技术,提出了常用的工业微生物对应底盘细胞的改造策略,以期为研究者在进行微生物底盘细胞改造时提供参考。
1 CRISPR/Cas9系统
CRISPR/Cas9系统由3个功能组分组成:crRNA(CRISPR RNA,crRNA)、tracrRNA(trans-activating CRISPR RNA,tracrRNA)及Cas9蛋白。三者结合为复合物,识别特定的NGG序列,即前间区序列邻近基序(protospacer adjacent motif,PAM)区,crRNA通过碱基互补配对与附近间隔序列作用,Cas9蛋白负责对特定靶标DNA进行双链断裂,在实际应用过程中crRNA和tracrRNA通常被结合在一起简化为小向导RNA(small guide RNA,sgRNA)[3]。图1a和1b分别展示了CRISPR系统组成及Cas9核酸酶功能结构域。不难发现该系统有两个重要特点:一是crRNA可以通过碱基互补配对原则精确识别基因组靶位点;二是Cas9蛋白可以对靶DNA进行切割,引发双链断裂。科学家正是直接或间接运用了这两点而发展出了以下的基因编辑技术。
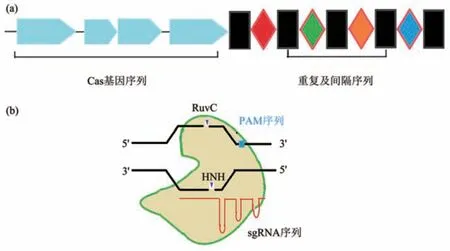
Fig. 1 The composition of CRISPR (a) and Cas9 nuclease functional domain (b)图1 CRISPR系统组成(a)和Cas9核酸酶功能结构域(b)
2 CRISPR相关的基因编辑技术
目前CRISPR相关的基因编辑技术在微生物底盘细胞改造中的应用具体表现在以下5个方面:CRISPR耦联的非同源末端连接(nonhomologous end-joining,NHEJ)修复系统、CRISPR耦联的同源定向修复(homology directed repair,HDR)系统、CRISPR耦联转录抑制因子/激活因子(CRISPR interference/activate,CRISPRi/CRISPRa)、CRISPR耦联脱氨酶单碱基编辑(base edit,BE)、CRISPR耦联转座。以下将对这5种CRISPR衍伸技术分别介绍。
2.1 CRISPR耦联NHEJ修复系统
NHEJ修复机制主要存在于高等真核生物中,修复的结果往往伴有宿主基因组核苷酸随机的插入或删除。因此NHEJ修复并不适用于对宿主基因组进行精确地编辑,NHEJ诱导的核苷酸序列改变却丰富了基因的多样性。
遗憾的是,大多数细菌缺乏天然的NHEJ修复机制,可能原因是在这些宿主中缺乏参与修复系统的Ku蛋白,该蛋白质通过结合断裂DNA末端,从而防止DNA降解。事实上,NHEJ系统存在于诸如分枝杆菌、芽孢杆菌和链霉菌等类群。山东大学的科研人员Su等[4]通过在大肠杆菌中异源表达来自于结核分枝杆菌(Mycobacterium tuberculosis)的Ku蛋白和LigD蛋白,重建出NHEJ途径,具体机制参见图S1a。通过该方法,研究者实现了大肠杆菌基因组3~17 kb长片段的删除。进一步地,Zheng等[5]通过将来自耻垢分枝杆菌(Mycobacterium smegmatis)的NHEJ修复机制引入大肠杆菌,实现了更长123 kb DNA片段的缺失。可见,NHEJ修复系统不依赖于模板及抗性标记,便可实现长片段删除,被广泛应用于宿主基因组的精简。表1列举了CRISPR耦联的NHEJ修复系统在各微生物细胞改造中的具体应用。

Table 1 CRISPR coupled NHEJ in microbial modification application表1 CRISPR耦联的NHEJ修复系统在微生物细胞改造中应用
2.2 CRISPR耦联HDR系统
上述提到NHEJ修复将导致基因组随机插入或缺失片段,与之不同的是HDR可实现基因组的定向改造。HDR的机制具体参见图S1b。
Jiang等[10]发现,CRISPR引发的双链断裂(double strand break,DSB)可以促进同源重组,该过程可以作为一种压力,筛选经同源重组而存活下来的宿主。通过进一步研究,Yang等[11]成功构建了一个大肠杆菌CRISPR/Cas9双质粒系统基因编辑平台,Cas9质粒表达的Cas9蛋白,与CRISPR质粒表达的sgRNA结合,引发基因组靶位点双链断裂。此外Cas9质粒表达的Exo、Beta、Gam 3种蛋白质协助与受体菌基因发生同源重组,最终实现了基因的断裂、重组修复和质粒的消除。Zhao等[12]将以上双质粒系统简化为单质粒,使得编辑过程更为简单,但对供体片段(donor DNA)长度有限制,且未能解决多位点同时编辑问题。Shukal等[13]通过不对称PCR构建的供体片段,避免了需重叠PCR的繁琐过程,简化了编辑的步骤。
目前 CRISPR辅助的同源重组已被用于开发多种高效一步无疤基因组工程编辑,其中涵盖传统工业微生物的编辑,如大肠杆菌、枯草芽孢杆菌、谷氨酸棒状杆菌、恶臭假单胞菌等。表2列举了CRISPR耦联的HDR系统在各微生物细胞改造中的具体应用。

Table 2 CRISPR coupled HDR in microbial modification application表2 CRISPR耦联的HDR系统在微生物细胞改造中应用
2.3 CRISPR耦联转录抑制/激活因子
Cas9蛋白包含2个核酸酶结构域,分别为RuvC和HNH,这2个结构域具有切割双链DNA的功能。当Cas9蛋白引入H840A和D10A突变时,其失去了内切酶活性,形成所谓的失活Cas9(dCas9)。dCas9蛋白虽不能切割DNA,但其仍具有协助指引RNA结合DNA的功能。Qi等[22]在大肠杆菌中研究了CRISPR/dCas9系统,发现其具有特异性抑制基因表达的能力,并发展成为CRISPRi技术。研究者普遍认为dCas9通过靶向启动子序列并阻断转录起始复合物而有效地沉默转录。与前述的两种改造底盘细胞代谢原理不同,CRISPRi系统无需诱导DSB,便实现了灵活的多路基因沉默,大大减少了代谢工程所需的时间。
当dCas9蛋白与一个转录激活子融合时,通过dCas9/crRNA特异性的DNA结合活性,激活子可以被间接地放置在目标基因的转录起始位点附近,并招募转录起始复合物从而在转录水平上增强靶基因的表达。随后该技术被称为CRISPRa。Bikard等[23]通过将RNA聚合酶亚基RpoZ融合到dCas9蛋白的C端,并靶向大肠杆菌MG1655基因组中的一个转录起始点上游的弱启动子序列,使得其转录活性增强了23倍。该团队还发现CRISPRa系统的激活效应在弱启动子中尤为显著,而对于强启动子的增强并不明显。与CRISPRi转录抑制相比,dCas9-ω复合物转录激活作用较为有限。Hu等[24]研究使用改进的噬菌体辅助连续进化技术(phageassisted continuous evolution,PACE),筛选获得的Cas9突变体,该突变体通过与ω亚基复合,使得转录激活效率增加了近100倍。表3列举了CRISPRi/CRISPRa在各微生物代谢途径编辑的具体应用。CRISPRi/CRISPRa 的作用机制参见图S1c和S1d。

Table 3 CRISPR coupled CRISPRi/CRISPRa in microbial modification application表3 CRISPR耦联的转录抑制/激活因子在微生物细胞改造中应用
2.4 CRISPR耦联脱氨酶单碱基编辑
碱基编辑器(base editor)是一种脱氨酶与受损的CRISPR/Cas核酸酶(无法切割DNA双链)融合蛋白,由Komor等[33]首先构建,常用于无模板基因编辑。受损的Cas核酸酶Cas9(SpCas9)可以是催化失活的dCas9,完全不能切割DNA双链,或一个Cas9 nickase(nCas9,保留D10A突变),破坏未编辑链。通过sgRNA的引导,融合蛋白被招募到靶位点,但不引起DSB,其中胞嘧啶脱氨酶与受损的Cas9融合(pyrimidine base editor,CBE)将胞苷C转化为胸腺嘧啶T,腺嘌呤脱氨酶与受损的Cas9融合(adenine base editor,ABE)将腺苷A转化为鸟苷G。Kurt等[34]将尿嘧啶-DNA糖基化酶(Ung)与受损的Cas9融合,在大肠杆菌中实现了胞苷C转化为腺苷A。碱基编辑器的具体机制参见图S2a和S2b。目前碱基编辑器主要用于传统工业微生物如大肠杆菌、谷氨酸棒状杆菌、酿酒酵母等基因的失活。Wang等[35]利用CBE对谷氨酸棒状杆菌中3基因odhA、pyk和ldhA的组合失活,从而提高了谷氨酸产量。Zhong等[36]通过使用ABE突变天蓝色链霉菌(S. coelicolor)中的actVB起始密码子,使得放线氧酰基酮(actinoperylone,ACPL)的产量提高。虽然关于编辑器的应用在代谢工程上的报道较为有限,但将来可能出现直接应用碱基编辑来改变一个启动子或其他转录调控因子,如操作子、增强子、激活子结合位点,或翻译元件包括核糖体识别位点(Shine-Dalgamo,SD)序列、起始密码子等,都将是极具有吸引力的基因表达调控策略。
2.5 CRISPR耦联转座
优化特定基因的遗传剂量使得目标产物高效表达是制备高性能生物催化剂的关键步骤。但是传统依赖质粒方法在控制基因剂量上并不理想,原因是质粒复制子和影响基因拷贝数的各种外部因素影响。因此,一个可编辑、快速、无标记、定点染色体多拷贝集成系统将是一个强大的编辑工具应用于工业微生物底盘细胞改造。染色体整合方法实现了精确基因剂量控制与重组基因稳定性。最近研究发现,基于转座子的CRISPR基因组编辑技术可以高效、准确地整合基因元素进入一个特定的位点,该过程并不涉及DNA双链的断裂。CRISPR耦联转座过程参见图2a和2b。
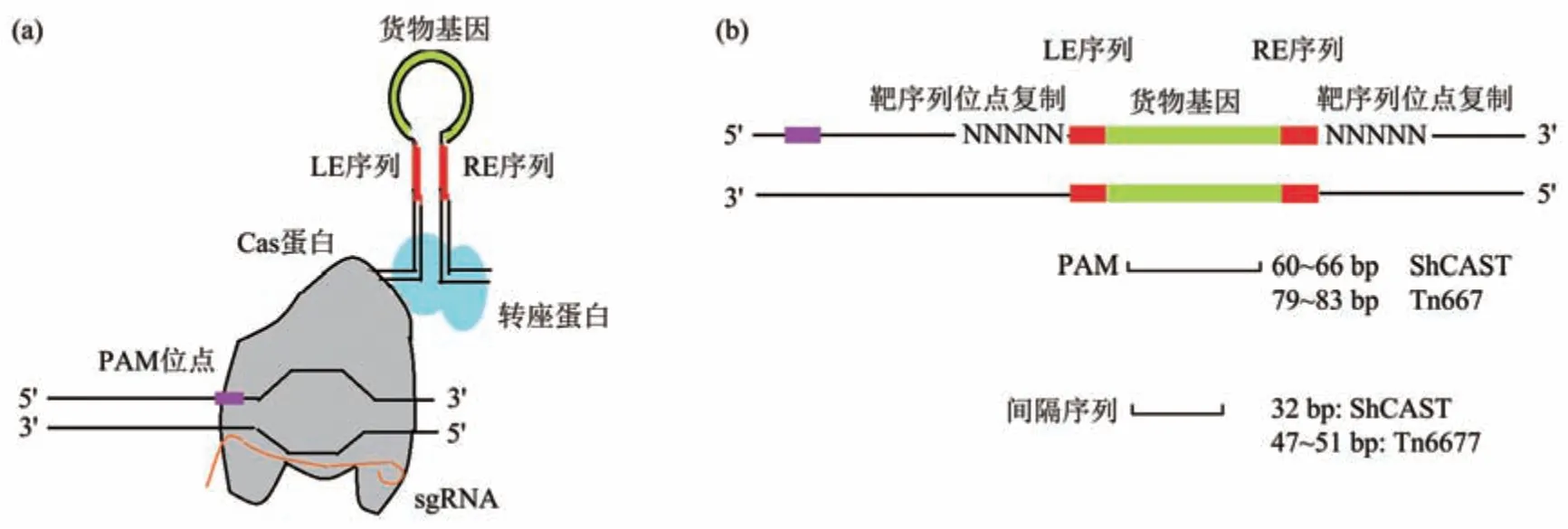
Fig. 2 Transposable process (a) and insertion genomic location (b)图2 转座过程(a)及插入基因组位置(b)
Zhang等[37]研究了CRISPR相关霍氏双岐藻(Scytonema hofmanni)转座系统ShCAST的特征。ShCAST蛋白复合物由一个基因集群编码的tn7样转位酶亚基(TnsB、TnsC以及TniQ)和Cas12k蛋白组成。TniQ和Cas12k识别目标位点并负责招募TnsC, 通过TnsB和TnsC间的相互作用,TnsB发挥裂解DNA的功能,将货物基因断裂,并将其插入位于PAM序列下游60~66 bp处。研究人员将该系统应用于大肠杆菌并成功插入了10 kb长片段。另一方面,Sternberg等[38]对来自霍乱弧菌的CRISPR相关转座酶进行了研究,发现该系统中Cascade蛋白复合物由Cas6、Cas7和Cas8组成,结合TniQ蛋白引导转座子将货物基因插入到靶目标基因组中。Yang等[39]比较了ShCAST和霍乱弧菌的向导RNA辅助靶向插入转座系统(insert transposable elements by guide RNA-assisted targeting,INTEGRATE),并总结出一套应用于大肠杆菌多基因同时敲入的CRISPR相关转座酶多拷贝染色体整合(multicopy chromosomal integration using CRISPR-associated transposases,MUCICAT),该系统由三质粒组成,通过设计8个间隔序列的微阵列,该团队将葡萄糖脱氢酶(GDH)基因同时整合到了BL21(DE3)基因组的8个位点,得到了一系列梯度拷贝的GDH基因,其中最优6个拷贝的菌株酶活是传统质粒pET24a-GDH发酵的2.63倍。本实验室通过将间隔序列靶向基因组的间隔序列(IS1),该IS1位点在BL21(DE3)基因组中存在29个拷贝,通过一轮转座便实现了GDH基因在IS1位点的插入。Sternberg等[40]将三质粒改进为一个单质粒系统,简化后的质粒,货物基因插入地方向一致,且长片段插入效率接近100%,进一步结合Cre-Loxp系统,通过同时靶向基因组的两个位点分别实现了2.4 kb、10 kb、20 kb长片段删除。
以上内容主要对5种编辑技术进行了单独的介绍,但应该指出的是,即使是对一种特定的底盘细胞改造,根据最终要达到的效果,中间编辑的过程也有可能需要多种编辑方法联合使用,如对基因组整合一个代谢途径而言,首先可以通过转座方法插入所涉及基因的长片段,然后借助同源修复系统去除因转座而存留在基因组两端的重复序列,最后出现的单碱基突变或启动子等元件的优化则用碱基编辑器进行细调更为合适,这样可能实现最短时间改造细胞的目的。随着CRISPR技术的迅猛发展,新的技术也是层出不穷,除以上描述的衍伸技术外,最近发展出的CRISPR耦连系统包括易错DNA复制、引导编辑、表观修饰等。虽然这些新工具还没有成熟,尚未应用于代谢工程,相信这些新技术必将丰富多个CRISPR调控系统在同一细胞中组合应用,使得代谢途径中基因的多重调控变得更方便高效。表4主要总结了以上CRISPR衍伸技术的特点。

Table 4 Comparison of CRISPR extension technology characteristics表4 CRISPR衍伸技术特点比较
上文主要介绍了微生物底盘细胞改造的具体技术,下文将对改造的对象——工业微生物底盘细胞以及CRISPR基因编辑方法如何在底盘细胞改造中具体应用进行重点描述。
3 CRISPR/Cas9在工业微生物底盘细胞改造中的应用
3.1 CRISPR/Cas9技术在大肠杆菌中的应用
大肠杆菌(Escherichia coli)因其培养方法简单、增殖周期短、耐受环境强的特点,被广泛用于工业发酵。Jiang等[10]在大肠杆菌中利用CRISPR/Cas9系统进行了基因组的编辑,研究者通过重组方法将一段突变序列替换了原始序列。Yang等[11]进一步利用CRISPR同源重组技术同时对大肠杆菌多至3个位点进行了基因敲除。除该技术外,CRISPRi系统也被用于大肠杆菌工业发酵,Wu等[41]利用CRISPRi抑制丙二酰辅酶A消耗通路,使得丙二酸盐前体物质积累,从而提高2S构型匹诺赛琳((2S)-pinocembrin)滴度至525.8 mg/L。除CRISPRi外,CRISPRa体系也于2013年在大肠杆菌构建成功。Bikard等[23]通过将dCas9的C端连入ω亚基,使得β半乳糖酶在转录水平上提高了2.8倍。可以看出CRISPR耦连HDR、CRISPR耦连转录激活/抑制因子,在大肠杆菌合成代谢途径中已有实际应用。
3.2 CRISPR/Cas9技术在谷氨酸棒状杆菌中的应用
谷氨酸棒状杆菌(Corynebacterium glutamicum)是一种革兰氏阳性土壤细菌,作为重要的微生物,过去常用于工业生产氨基酸,现已被改造合成各种化合物,包括高分子亚基、医药中间体和生物燃料等。目前已开发的谷氨酸棒状杆菌基因编辑系统主要集中在同源重组介导的CRISPR/Cpf1和CRISPR/Cas9技术。新凶手弗朗西丝菌(Francisella novicida)来源的CRISPR/Cpf1系统,由于其中的Cpf1相对于Cas9蛋白分子质量小,细胞毒性较低,利于构建到载体中提高转化效率,因此该系统被应用于较难编辑的谷氨酸棒状杆菌。2017年,杨晟团队[18]在谷氨酸棒杆菌中成功建立CRISPR/Cpf1系统,研究者将Cpf1蛋白和CRISPR RNA构建在同一个质粒中,以单链DNA为供体,实现了近90%编辑效率。CRISPR/Cas9系统方面,Liu等[42]采用双质粒共转方法以减少Cas9质粒在宿主中复制机会,避免Cas9质粒突变而产生假阳性。此外,研究者还将Cas9和gRNA质粒分别采用更为严谨的强启动子Ptac和P11F(一种PcspB衍生启动子)优化,优化后的系统在分别以质粒骨架和单链DNA(single strand DNA,ssDNA)为供体时,达到60%和80%的编辑效率。为实现更长片段的删除和插入,Wang等[19]先将Cas9整合入宿主基因组,单转化gRNA质粒以此提高转化效率,研究者发现,相对于载体形式表达Cas9蛋白,基因组表达的Cas9对宿主毒性更低,更有利于菌体生长。通过该方法作者实现了长20 kb片段删除以及7.5 kb片段插入,进一步通过对丙二醇合成途径相关基因改造,编辑后的菌株实现了丙二醇6.75 g/L产量。
3.3 CRISPR/Cas9技术在枯草芽孢杆菌中的应用
枯草芽孢杆菌(Bacillus subtilis)是革兰氏阳性菌芽孢杆菌的模式菌株,具有强大的蛋白质表达系统。目前,已在枯草芽孢杆菌中建立了CRISPR/Cas9、CRISPRi和CRISPR/Cpf1等编辑系统。Liu等[43]在枯草芽孢杆菌中应用CRISPRi系统,实现了乳糖-N-新四糖(LNnT)在摇瓶产量中达到2.30 g/L。Altenbuchner等[44]利用穿梭质粒在枯草中诱导表达Cas9蛋白,耦连HDR系统实现了amyE基因的25.1 kb长片段缺失。Wu等[45]通过CRISPR/Cpf1系统实现双精度框内基因敲除、多点突变。 随后利用该系统研究者在枯草芽孢杆菌中实现了N-乙酰氨基葡萄糖的生物合成途径。
3.4 CRISPR/Cas9技术在酵母菌中的应用
酵母是单细胞真核生物,由于其清晰简单的遗传学背景以及易操作性与安全性,常被用于工业发酵。2015年,Horwitz等[46]在酿酒酵母应用CRISPR/Cas9系统将6个基因片段共24 kb整合到基因组,建立了莽草酸合成途径。在基因敲除而实现代谢流调控方面,Xu等[47]在酵母中运用Cas9及优化后的sgRNA,对酵母基因组进行了4个位置基因突变并成功构建了乙醇耐受型酵母菌株。以上实例都是基于CRISPR/Cas9耦连同源重组在有供体DNA情况实现基因的敲入或敲除,可见同源重组修复系统用途之广。除此之外,dCas9也被用于酵母基因组转录水平调控。Gilbert等[48]通过dCas9抑制TEF1启动子的转录,从而降低基因的表达丰度。
对于利用CRISPR耦连转座系统在酵母中实现长片段基因插入实例尚未报道,但已有科研人员提出开发真核生物转座子系统或细菌Tn7-CRISPRCas系统从而实现DNA长片段的插入、多个基因控制的通路插入以及多个重要性状的叠加等。
4 总结和展望
CRISPR技术的出现极大方便了微生物底盘细胞改造,但目前CRISPR技术还面临以下问题。
a. PAM序列的依赖性。目前的CRISPR/Cas9技术由于NGG序列限制,尚不能对任意位点编辑。CRISPR技术普适性的应用,需要打破现有PAM序列,在识别位点上加以拓宽。
b. 编辑的脱靶率。对于减弱脱靶效率,目前研究者们已通过结构优化或者随机突变的形式获得高保真的突变体。但是这些突变体打靶效率通常较低,因此,在保持目标编辑活性的同时提高Cas9特异性对于探索高保真和高效的Cas9工具至关重要。Sun等[49]最近在Cas9基础上通过对7个残基突变,得到一种新型SuperFi-Cas9,该突变蛋白质裂解靶DNA的速度与野生型相当,但具有更高的脱靶识别性能,SuperFi-Cas9的裂解活性只在体外进行了测试,临床应用还需要进一步研究,继续优化发展真正意义上的高保真突变体将是未来的研究重点。
c. 应用广泛性有待提升。虽然CRISPR技术已经广泛应用于多种微生物,但在某些微生物中依然存在Cas9蛋白毒性问题而难以应用,虽然Cpf1可以缓解但依然存在较低毒性。
从底盘细胞构建上看,该过程涉及大量基因的敲除,这首先要求对基因信息进行注释,以便决定编辑的位置。基因信息的注释需要生物信息学分析,过程较为复杂。此外,尽管目前的模式微生物代谢特征很好,而且已有多种工具可对其基因快速修饰,在代谢工程中已取得广泛的应用,然而这些模式菌株仍是无法生产所有理想的产品,以满足日益增长的工业需求。相比之下,来自更复杂环境的非模式微生物已进化到具有利用更便宜、更环保的碳源完成多种生理功能和代谢的能力,这对未来生产生物燃料以及药物和新型抗生素都很重要。非模式微生物能忍受极端工业加工环境,如低pH、高盐、高温,已然成为代谢工程研究的热点之一。然而,由于缺乏简单的基因编辑工具,阻碍了非模式微生物基因的表征和表达修饰。相信未来科学家根据不同微生物特点而开发出相应的编辑工具,用于更多产品生产。这些问题的解决必依赖于CRISPR技术的革新,在微生物底盘细胞改造方面取得更加广泛应用,从而实现更多附加值产品的底盘细胞。
附件见本文网络版(http://www.pibb.ac.cn或http://www.cnki.net):
PIBB_20220323_Fig S1.tif
PIBB_20220323_Fig S2.tif
