嵌合抗原受体巨噬细胞免疫疗法及其在实体瘤治疗中的应用*
2023-07-19杨艳甘妮翠彭宁馨张岚萱张继虹李锦媛
杨艳 甘妮翠 彭宁馨 张岚萱 张继虹 李锦媛**
(1)昆明理工大学医学院,昆明 650500;2)云南省第一人民医院普外一科,昆明 650032)
随着肿瘤免疫疗法研究的逐渐深入,以过继性嵌合抗原受体T细胞(chimeric antigen receptor T-cell,CAR-T)免疫疗法、免疫检查点抑制剂为代表的多种免疫治疗策略相继被开发并进入临床应用[1]。然而当前的免疫疗法主要针对适应性免疫,CAR-T在实体瘤中浸润不足,疗效并不理想[2-3]。与此同时,研究发现先天免疫细胞也确实具有发挥抗肿瘤活性的潜力[4]。巨噬细胞是实体肿瘤微环境中主要的免疫浸润细胞,可吞噬肿瘤细胞,并将肿瘤抗原呈递给T细胞进而启动肿瘤特异性免疫应答[5-6]。因此,改造巨噬细胞成为嵌合抗原受体巨噬细胞(chimeric antigen receptor macrophage,CAR-M)代表了一种新型抗肿瘤固有免疫疗法,可与放疗或者针对适应性免疫的治疗方法协同作用,发挥更强劲的抗肿瘤免疫作用。本文主要介绍CAR-M的发展历程、结构设计和临床试验相关研究进展,以期为其未来的临床应用提供参考。
1 嵌合抗原受体(CAR)修饰的免疫细胞用于肿瘤免疫治疗
21世纪初,靶向T细胞活化的相关免疫疗法就已启动,CAR-T细胞是通过基因工程技术制备的抗原受体T细胞,可与肿瘤细胞表面的抗原特异性结合,具有效应性T细胞的T细胞受体(T cell receptor ,TCR)特异性和非主要组织相容性复合体(major histocompatibility complex,MHC)限制性抗肿瘤活性的特点。CAR的主要结构涉及识别抗原的单链抗体可变区片段(single chain antibody variable fragments,scFvs)、跨膜区、TCR β链CD3ζ(T细胞活化第一信号)和协同刺激信号结构域(T细胞活化第二信号)(图1)[7-8]。 虽然CAR-T细胞治疗血液肿瘤的临床效果良好,但由于实体肿瘤细胞中抗原缺失、缺乏特异抗原以及免疫抑制肿瘤微环境等原因,其对实体瘤的治疗效果并不理想。同时在临床上还发现,CAR-T细胞疗法会产生细胞因子释放综合征(cytokine release syndrome,CRS)、神经方面的相关不良反应[3]。由此在完善CAR-T免疫疗法的同时,拓宽CAR策略也非常必要。目前CAR策略已经用于修饰自然杀伤细胞(natural killer,NK)、γδT细胞(gammadelta T cells ,γδT)、自然杀伤性T细胞(natural killer T cells,NKT)、调节性T细胞(regulatory T cells,Treg)等免疫细胞,研究表明通过对以上免疫细胞进行CAR修饰对肿瘤免疫治疗具有积极作用[9-13]。
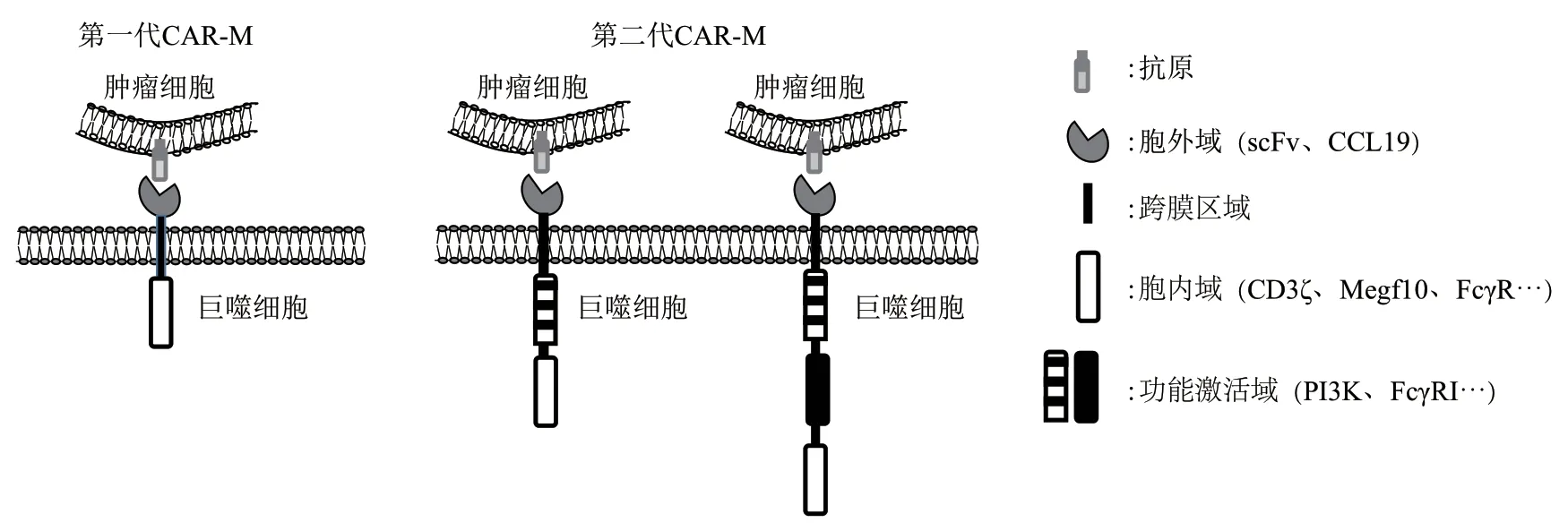
Fig. 1 Structure display of CAR-M图1 CAR-M结构示意图
肿瘤微环境(tumor microenvironment,TME)由多种细胞、细胞因子、酶类以及胞外基质共同组成[14]。大量活化的巨噬细胞浸润肿瘤间质,被称为肿瘤相关巨噬细胞(tumor-associated macrophage,TAM),在大多数实体肿瘤的TME中占比达50%[15-16]。除此之外,巨噬细胞还能够分泌基质金属蛋白酶(matrix metalloproteinase,MMP),从而破环肿瘤细胞外基质层进入到肿瘤内部发挥作用[17-18]。巨噬细胞的这一特质推动了利用巨噬细胞治疗肿瘤的研究。然而研究人员发现,直接进行巨噬细胞移植治疗效果甚微[19-21]。其主要问题在于,TME能够诱导巨噬细胞分化为替代活化型 (M2表型),M2型巨噬细胞能通过分泌多种活性物质促进血管生成、肿瘤细胞增殖、转移、免疫抑制、免疫逃逸和耐药性等[22-23]。研究证明,被极化为M2表型的巨噬细胞可以在一定程度上逆转为M1表型[24]。M1型巨噬细胞高表达炎症细胞因子,具有较强的抗微生物和抗肿瘤活性。由此,逆转M2表型在肿瘤治疗中具有重要意义。同时,研究发现TME中的巨噬细胞需要额外的信号来引导其对抗肿瘤[25]。研究人员从CAR-T的成功中获得启示,通过转基因手段促使巨噬细胞表达靶向特定肿瘤抗原的CAR转变为CAR-M,从而增强或激活其对目标肿瘤细胞的吞噬杀伤作用[26]。由于巨噬细胞在TME中大量存在,且能够进入肿瘤内部发挥作用,因此CAR-M也成为针对实体肿瘤最有希望的临床免疫疗法之一。
2 CAR-M的设计与应用
最初,CAR的原始结构是为T细胞设计的,没有专门为巨噬细胞设计的CAR结构。CAR-M结构参照CAR-T进行设计,主要由识别特定肿瘤抗原的细胞外信号域、跨膜区域和细胞内结构域组成(图1)。后续在CAR-NK相关研究中发现,CAR结合表位的位置及其与细胞表面的距离可影响抗原结合和细胞活化[27]。第一代CAR-M主要对CAR的细胞外结构域进行改造,以靶向特定的抗原来识别和吞噬肿瘤细胞。目前,通过对细胞外信号域的研究,确定了几种常见的肿瘤靶点,如CD19、CD22、HER2等[28]。
目前,第二代的CAR-M正在开发中,除了保持第一代CAR-M技术的特点外,研究人员希望可以通过在CAR结构中添加一个胞内结构域来实现肿瘤相关抗原呈递和T细胞活化等目标。加利福尼亚大学的Morrissey团队[26]制备了一种名为嵌合抗原吞噬受体(chimeric antigen receptors for phagocytosis,CAR-P)的新型CARs,并将其引入巨噬细胞中。该团队从已知的小鼠吞噬受体库中筛选出Megf10、FcγR作为细胞内结构域以促进巨噬细胞的吞噬作用。后续实验证实,携带Megf10(CAR-PMegf10)或FcRV(CAR-PFcγR)细胞内结构域的CAR-P转染巨噬细胞后其对CD19的吞噬作用显著增强。这说明CAR-PMegf10或CARPFcγR结构域可促进巨噬细胞对特定配体靶标的吞噬作用。已有研究表明,PI3K信号通路对于大靶点的内化非常重要[29]。由此,该团队在CARPMegf10的结构基础上串联PI3K招募域,发现PI3K招募域的加入进一步增强CAR-M对癌细胞的吞噬。上述研究表明,将各类促吞噬效应因子的活化基序串联至CAR胞内结构域可促进CAR-M的吞噬活性和抗肿瘤效应。这可以作为一种应用于临床治疗癌症的思路,但这种策略需要对CAR-P受体本身进行更多优化,技术和成本方面依然面临较大的挑战。
为了使CAR-M获得更广谱的抗癌功效,除了CAR-M的结构,CAR基因的转染方法也需要考虑。在过去的CAR转染策略中,多以慢病毒作为载体对体内免疫细胞进行基因编辑,但其是否可以安全地用于临床治疗一直饱受争议[30]。为了更高效地转染原始人类巨噬细胞,宾夕法尼亚大学的Klichinsky团队[31]设计了一种复制缺陷型嵌合腺病毒载体(Ad5f35),并将抗HER2的CAR克隆到Ad5f35载体中,实验证明HER2-CAR-Ad5f35转染的人巨噬细胞高效表达了HER2-CAR,多呈现经典激活途径的M1型。同时腺病毒转染不影响巨噬细胞表面趋化因子受体的表达和迁移能力。为突破限制CAR-M体外大规模生产的技术瓶颈、高额费用和病毒载体修饰体内巨噬细胞导致的致癌可能性。Kang等[32]利用非病毒载体,对TME中的巨噬细胞进行体内修饰以治疗实体肿瘤。该团队首先将CAR-γ干扰素(IFN-γ)抗ALK基因克隆到非病毒载体piggyBac转座子中,得到CAR-IFN-γ载体(CAR-IFN-γpDNA)。随后利用一种22 ku大小且带正电荷的转染试剂PEI的衍生物——jetPEI-macrophage (MPEI)与带负电荷的CAR-IFN-γpDNA通过静电作用形成纳米复合物 MPEI/pCAR-IFN-γ,该复合物对人巨噬细胞的平均转染效率为14%。对比病毒载体而言,MPEI/pCARIFN-γ转染复合物更加简单实惠,但仍需进一步提高其转染率(表1)。

Table 1 Characteristics of different transfection vectors for CAR-M表1 CAR-M不同转染载体的特点
3 CAR-M的抗肿瘤效应
目前,CAR-M研究的重点是通过引入各类胞内致敏结构域激活和增强巨噬细胞的吞噬作用,以促进CAR-M发挥抗肿瘤作用(表2)。
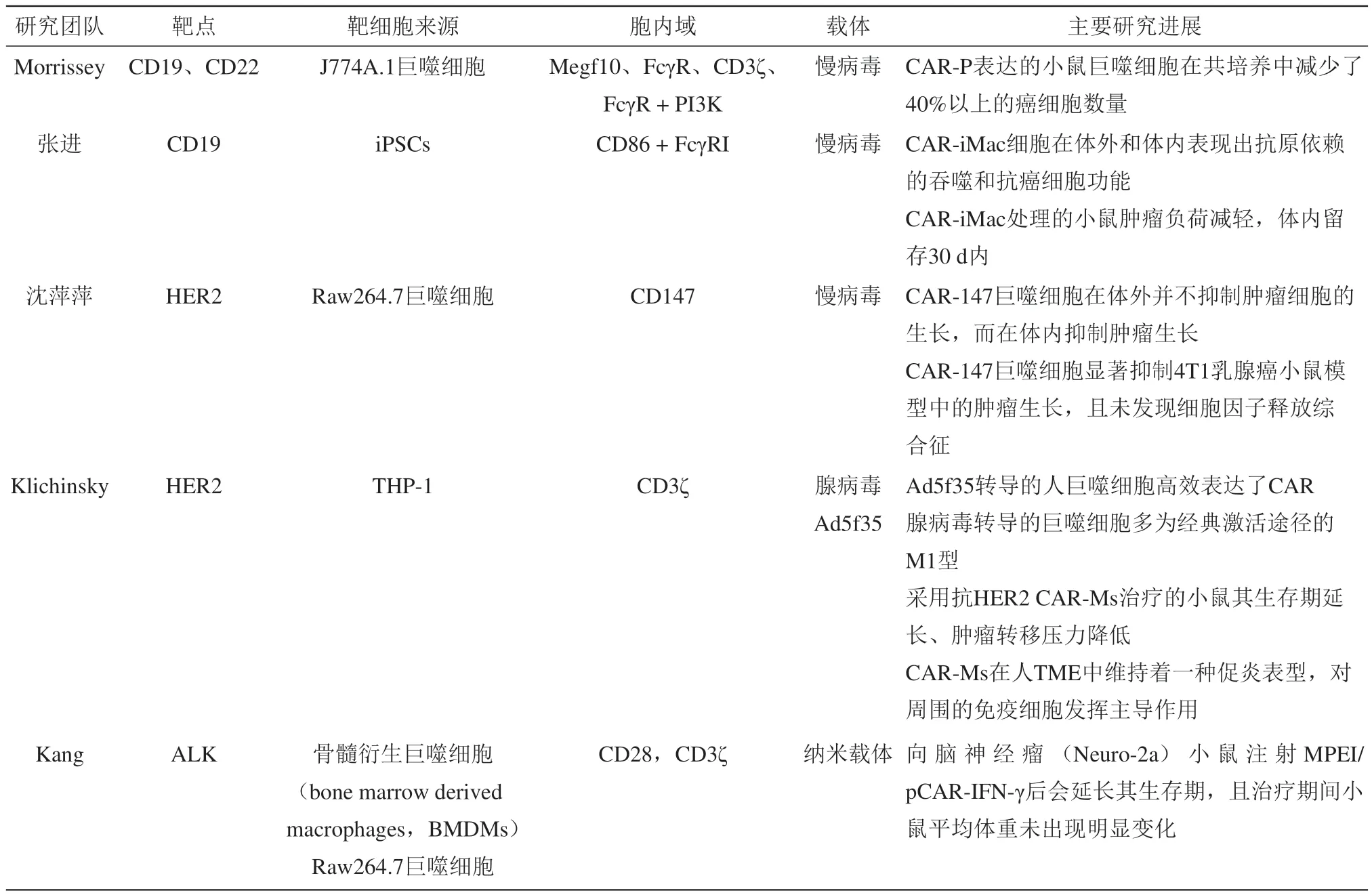
Table 2 The anti-tumor effect of CAR-M表2 CAR-M抗肿瘤研究进展
加利福利亚大学的Morrissey团队[26]制备的CAR-P巨噬细胞与人Burkitt’s淋巴瘤细胞Raji B共培养44 h后,发现CAR-P巨噬细胞的加入显著减少了Raji B细胞的数量。但该研究没有进一步探究Raji B细胞的减少主要是由巨噬细胞的吞噬效应介导还是巨噬细胞诱导的肿瘤细胞自噬性死亡。总的来说,CAR-P是一种成功的策略,可以引导巨噬细胞作用于肿瘤靶细胞,并可以启动细胞吞噬和自噬作用,从而清除肿瘤细胞。
浙江大学张进团队[33]从健康供者的外周血中分离得到诱导性多能干细胞(induced pluripotent stem cells,iPSCs),将促进M1型极化的CARs稳定转染到iPSCs中,继而诱导其分化为M1-CAR-iMacs。体外肿瘤细胞杀伤实验进一步证实M1-CAR-iMacs的抗肿瘤作用及抗原依赖性的M1极化能力。在荷瘤NSG小鼠模型中,CAR-iMac细胞在体内可以持续存活20 d以上,30 d后逐渐消失。特异靶向肿瘤间皮素的CAR(meso)-iMac处理荷瘤小鼠后第4、11和14天肿瘤负担持续减轻,这说明CAR(meso)-iMac在体内具有抗癌细胞活性,但是需要进一步改进CAR-iMac以维持其活性。
南京大学沈萍萍研究团队[34]设计了一种靶向肿瘤细胞外基质的嵌合抗原受体巨噬细胞(CAR-147),CAR-147可特异性识别抗原HER2,并有效激活巨噬细胞中MMP的表达。CAR-147巨噬细胞在体外并不抑制肿瘤细胞的生长,而在体内抑制肿瘤生长。体内实验中CAR-147巨噬细胞可降解肿瘤周围致密的胶原基质,抑制肿瘤转移,促进肿瘤模型中T细胞的浸润。同时,CAR-147巨噬细胞显著增加肿瘤组织中细胞因子IL-12和IFN-γ的表达水平。
宾夕法尼亚大学的Klichinsky团队[31]将特异性识别HER2的CAR克隆到Ad5f35载体中,转染巨噬细胞得到HER2-CAR-Ms。体外实验证明,HER2-CAR-Ms对HER2阳性微珠和肿瘤细胞具有特异性吞噬作用,而且其对肿瘤的吞噬和杀伤水平与CAR的表达水平直接相关。小鼠体内实验显示,接受HER2-CAR-Ms治疗的小鼠生存期延长、肿瘤转移率降低。CAR-M在无瘤NSGS小鼠体内存在至少62 d。且CAR-Ms在人TME中维持着一种促炎表型,可调控周围免疫细胞。除了直接抗肿瘤,HER2-CAR-Ms还可交叉提呈抗原和激活T细胞。这也证实了CAR-Ms直接减轻肿瘤负担、塑造TME和产生疫苗效应。该团队正在开展首次CAR-M的人类临床试验。
韩国首尔国立大学的Kang团队[32]使用纳米颗粒载体,将编码CAR和IFN-γ的基因转染巨噬细胞得到MPEI/pCAR-IFN-γ。脑神经瘤(Neuro-2a)荷瘤小鼠瘤内注射MPEI/pCAR-IFN-γ后生存期显著延长,且治疗期间小鼠平均体重未出现明显变化,提示MPEI/pCAR-IFN-γ治疗可以抑制肿瘤生长且副作用轻微。
4 总结
2021年9月,美国食品和药物管理局(FDA)给Klichinsky团队设计的CAR-M(CT-0508)开通快速审批通道,表明了对基于CAR设计的肿瘤免疫疗法的迫切需求[31]。尽管各种CAR-M设计的肿瘤免疫疗法在动物实验中的表现优良,但仍有许多问题需要解决。首先,体外改造后的巨噬细胞注射进入机体后不会增殖,不仅细胞耗费较大,而且疗效也因此受限。其次,转染CAR进入巨噬细胞的方法,也存在较多的技术限制,如载体的选择和长期安全性[35]。虽然表达CARs的病毒载体可以大量生产,并可在-80°C下保存约9年,但是载体的安全性、无菌性、效价、纯度至关重要[36-38]。另外,病毒载体的DNA通过插入突变整合到宿主细胞也是一个令人担忧的问题,目前开发的病毒载体较之前载体大大降低了突变的风险。然而,慢病毒和逆转录病毒载体都有潜在的致癌作用。非病毒载体不存在细胞毒性,但其最小有效剂量需要后续的临床研究证实[32]。更关键的是其转染率较低,在未来仍需攻克这一难题。同时,TME组成呈现复杂性和异质性,当前的CAR-M产品在动物模型中得到良好的效果,但对于人的临床效果未知[39]。根据以往CAR设计的经验,靶抗原的暴露不够充分可能导致CAR-M的治疗效果不如预期[28]。最后,巨噬细胞在体内的迁移特性也可能导致CARM策略治疗效果的不佳[40]。外源性的巨噬细胞一般先通过肺,再在肝脏中富集[41]。过去的CAR相关免疫治疗也表明,大多数靶向肿瘤抗原的免疫细胞倾向于在健康组织中集聚,导致潜在的脱靶毒性,导致功效受限,甚者产生毒性伤害机体[42-45]。在未来的临床治疗中,应最大限度地提高CAR-M的有效性和安全性。
