心衰进程中非编码RNA对线粒体功能的调控作用*
2023-07-19王硕赵云罡
王硕 赵云罡
(天津体育学院天津市运动生理学与运动医学重点实验室,天津 301617)
心衰是一种复杂的心脏疾病,可由多种不同的心脏疾病引起,如心梗、病理性或先天性心肌肥大、缺血性心脏病、高血压等。心脏作为高耗能器官,其所需ATP大约有95%来源于线粒体氧化代谢[1]。因此,正常的线粒体功能对于维持心肌细胞能量供应和正常生命活动具有重要意义。而且,线粒体作为一种特殊的含有双层膜结构的细胞器,不仅通过氧化代谢产生ATP,还参与细胞生长与适应、钙离子转运、炎症反应、细胞凋亡与自噬等多种生命活动[1]。此外,在触发细胞死亡中,线粒体也扮演了重要角色。在各种理化因素刺激或病理状态下,线粒体钙超载和活性氧(reactive oxygen species,ROS)增加会导致线粒体膜通透性转换孔开放,造成线粒体膜电位丧失,进而导致ATP产生受阻、细胞色素c等蛋白质释放,并通过坏死或凋亡通路触发细胞死亡[2-3]。线粒体功能障碍诱导的细胞死亡被认为是心衰形成的重要机制,涉及线粒体氧化还原与蛋白质修饰变化、线粒体钙失衡、线粒体能量代谢变化、线粒体氧化应激、线粒体相关炎症反应等[1]。随着深度测序技术的发展,大量非编码RNA(non-coding RNA,ncRNA),包括微小RNA(micro RNA,miRNA)、长链非编码RNA(long non-coding RNA,lncRNA)、环状RNA(circular RNA,circRNA)等,被证实在基因转录和翻译调控过程中发挥重要作用,一些ncRNA还可以编码小肽。研究表明,ncRNA可通过调节靶基因表达影响心肌细胞增殖与分化、心肌重塑、心肌肥大、心肌纤维化等多种心脏生理病理活动[4-5]。ncRNA还可通过调节靶基因或靶蛋白影响线粒体结构与功能进而调控心脏病理生理变化,影响心衰进程,为心血管疾病诊断与治疗提供了众多新的研究靶点和思路。因此,探讨心衰发生和发展过程中ncRNA调控线粒体的作用机制,对开发心血管疾病分子诊断和靶向治疗新技术新手段具有重要意义。
1 线粒体与心衰概述
线粒体对于细胞存活至关重要,其广泛参与各种细胞活动。线粒体的主要功能之一是通过氧化磷酸化产生ATP。此外,线粒体还可产生ROS、氧化还原分子以及多种生物分子合成所需的中间物质[6]。ROS是氧化磷酸化的正常产物,可作为信号分子参与多种生理过程,但ROS过量产生会诱发氧化应激,对机体产生不良影响。过量ROS会造成蛋白质和脂质过氧化、DNA损伤、细胞功能障碍,甚至不可逆转的细胞损伤和死亡,这些反应促进了病理性心血管疾病的发生发展,特别是氧化应激反应增强与心衰过程中心肌重塑的病理变化密切相关[7-8]。
在心衰发展过程中,心肌细胞能量代谢会发生显著变化。正常生理状态下,心肌细胞所需能量主要来自线粒体氧化磷酸化,约占95%,主要以线粒体脂肪酸、葡萄糖和乳酸为底物,少部分来自糖酵解,约占5%;而在心衰过程中,心肌代谢发生重塑,脂肪酸氧化减弱,糖酵解代谢增强,供能水平下降,并造成乳酸增多[1,9-10]。脂肪酸氧化能力下降主要是由衰竭心肌线粒体功能障碍造成的线粒体脂肪酸氧化相关酶的表达及其活性下降导致的[11]。而心肌能量供应水平下降又会导致氧化磷酸化解耦联,进而引起线粒体ROS增加,产生氧化应激[12]。糖酵解反应增强还会造成细胞质H+积累,使Na+/H+交换增加,进而诱导Na+/Ca2+交换增加,造成胞质Ca2+聚集形成钙超载[13]。值得注意的是,ROS增多和钙超载是导致线粒体膜通透性转换孔开放的重要因素,膜通透性转换孔的开放一方面会促使线粒体内物质释放,如细胞色素c,进而促进细胞凋亡,另一方面也会导致膜电位丧失,影响ATP合成,最终造成细胞功能障碍[1]。另外,衰竭心肌的线粒体蛋白修饰也会发生变化,如蛋白质乙酰化水平明显增加,包括底物氧化相关蛋白酶在内的大量线粒体蛋白会在心衰进程中乙酰化,如丙酮酸脱氢酶、脂肪酸氧化酶、三羧酸循环相关酶等[14],也有研究表明,蛋白质氧连接的N-乙酰葡糖胺修饰过度,也会造成心衰[15]。
钙平衡失调是促进心衰发展的另一重要标志。钙离子是协调线粒体氧化还原与兴奋收缩耦联的重要第二信使[16],其可以激活多种线粒体代谢酶,如丙酮酸脱氢酶、异柠檬酸脱氢酶、α酮戊二酸脱氢酶等,并在调节氧化磷酸化、ROS清除及线粒体膜通透性转换孔开放相关蛋白中扮演重要作用[1,16]。肌质网钙回收受损与经由利阿诺定受体的钙泄漏导致收缩期胞质钙瞬变减少,造成心肌收缩功能障碍[11]。线粒体动力学变化也具有重要的病理生理调控意义。在不同类型细胞中或不同应激条件下,线粒体表现出不同的形态、大小、连接性及代谢状态,对维持细胞稳态至关重要。在病理条件下,线粒体融合与分裂失衡,导致碎片化的或超融合的线粒体发生聚集,影响能量代谢、氧化还原、钙平衡等过程,最终造成细胞死亡[16-18]。
在心衰发生与发展过程中,炎症反应增加也是不可忽视的重要因素。转录组学分析显示,心衰人体心脏和非心衰人体心脏相比,固有免疫反应相关基因的表达谱具有显著差异[19]。炎症反应的发生在心衰中的作用机制虽还不十分明确,但炎症反应会导致左室功能障碍和左室重构,且与心肌损伤密切相关,进而对心衰产生重要影响[20]。心衰过程中的心肌重塑涉及心肌肥大、心肌纤维化、细胞外基质重塑等,系统性炎症的发生则会促进心肌肥大和纤维化,系统性炎症主要由模式识别受体介导的固有免疫反应所触发,如NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor thermal protein domain associated protein 3,NLRP3)、Toll样受体4(Toll-like receptors,TLR4)等介导的固有免疫反应[12]。
2 心衰进程中ncRNA调控线粒体功能
2.1 miRNA对心衰进程中线粒体功能的调控作用
miRNA是一种内源性的单链短RNA,大小约为22个核苷酸。在经典的miRNA合成途径中,原始miRNA经由RNA聚合酶转录产生后,由Drosha复合体处理形成前体miRNA,并被输出蛋白运送到细胞质,在此进一步经由Dicer复合体切割形成成熟miRNA[21]。miRNA主要通过结合信使RNA(messenger RNA,mRNA)的3'非编码区(3' untranslated region,3'UTR)调节靶基因表达[22]。近年来,众多研究证实miRNA对线粒体结构与功能调控发挥了重要作用。大量miRNA在胚胎期、出生后及成年期心脏均有表达,且表现出不同的表达水平,这些miRNA的表达水平变化或缺失与心肌细胞分化异常、心脏发育紊乱、心脏功能障碍等病理变化的发生密不可分[23]。研究显示,心衰患者血清中,多种miRNA水平异常,如miR-122、miR-210、miR-423-5p、miR-499和miR-622等,这些miRNA有望成为心衰诊断和预后预测的标志物[24]。miRNA可通过多种通路调控线粒体功能改善心衰进程(表1)。
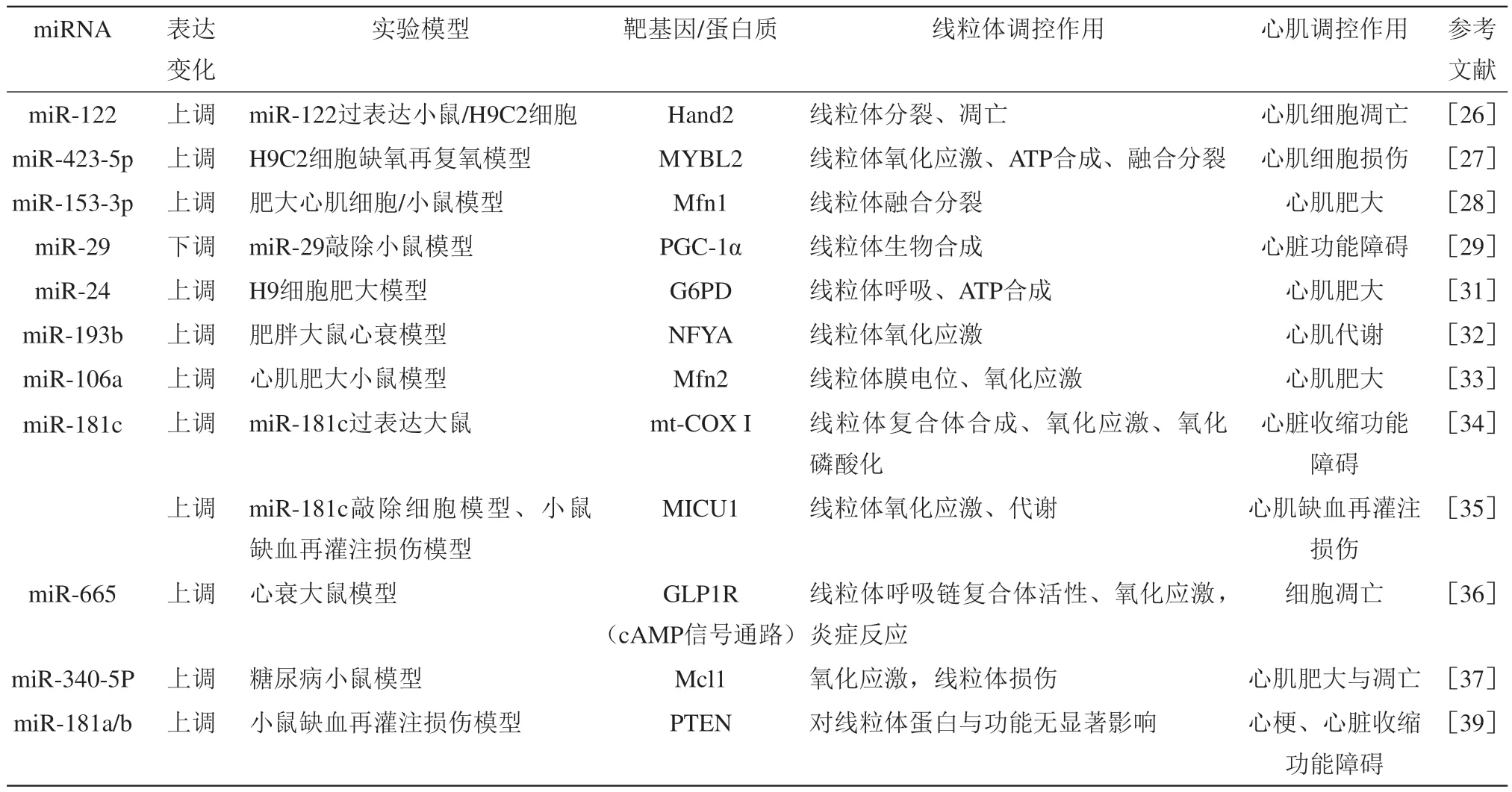
Table 1 Regulatory effect of miRNA on mitochondrial function and its mechanism during heart failure表1 miRNA对线粒体功能的调控及影响心衰进程的机制
线粒体融合分裂对于维持其正常功能至关重要,在应激条件下,正常线粒体可以通过融合损伤线粒体缓和刺激,线粒体分裂则是新线粒体产生和线粒体质量控制所必需的,且有助于在细胞受到强烈刺激时,通过线粒体分裂移除损伤线粒体,促进损伤细胞凋亡[25]。miR-122是心衰患者中高表达的一种miRNA,可靶向抑制转录因子Hand2,促进调节线粒体分裂的动力相关蛋白1(dynaminrelated protein 1,Drp1)的表达,诱导线粒体分裂和细胞凋亡,促进心衰发生[26]。另一种在心衰患者中异常高表达的miR-423-5p,可通过靶向调节Myb相关蛋白B(Myb-related protein B,MYBL2)的表达造成线粒体膜电位丧失、ATP减少、ROS生成和 Drp1表达增加,最终促进细胞凋亡;而抑制miR-423-5p可激活Wnt/β-catenin信号通路,改善线粒体功能障碍,减少细胞凋亡,从而减轻缺氧刺激造成的心肌细胞损伤[27]。在异丙肾上腺素(isoprenaline,ISO)诱导的心肌肥大模型中,miR-153-3p表达上调,并特异性降低线粒体融合蛋白1(mitofusin 1,Mfn1)的表达,导致线粒体分裂增加和心肌肥大,促进了线粒体碎片化和心肌纤维化;敲低miR-153-3p可以改善心肌线粒体碎片化和心肌肥大[28]。同时,活化T细胞核因子异构体c3(nuclear factor of activated T cells,NFATc3)作为miR-153-3p上游激活剂,可与miR-153-3p启动子结合,促进其转录表达,调节线粒体分裂和心肌细胞肥大。miR-29在多种纤维化疾病中表达下降,miR-29缺失会导致小鼠出现血管重塑和系统性高血压,形成射血分数保留的心衰,并表现出代谢紊乱、心肌纤维化、收缩功能降低等病理变化;敲除miR-29会使其靶基因过氧化物酶体增殖物激活受体辅助激活因子1α(peroxisome proliferator-activated receptor-gamma coactivator 1 alpha,PGC-1α)表达增加,造成线粒体生物合成加剧、线粒体密度增加、尺寸变小,心肌肥厚,代谢改变,糖酵解水平增强;而敲除miR-29的同时对PGC-1α进行单突变,可改善线粒体形态变化和心功能[29]。
氧化应激是促进心衰发展的重要因素,其可通过影响细胞代谢、促进细胞死亡、纤维化、炎症等多种病理变化促进心肌病变和功能障碍[30]。研究显示,miR-24可通过调节葡萄糖6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)表达,改善肥大心肌细胞线粒体功能,降低氧化应激[31]。在苯肾上腺素诱导的肥大心肌细胞模型中,miR-24可直接绑定G6PD的3'UTR,抑制其表达,而抑制miR-24可恢复G6PD表达,逆转肥大心肌细胞的线粒体功能障碍,提高线粒体呼吸链复合体活性和ATP浓度。在保留射血分数的心衰大鼠模型中,肥胖大鼠表现出代谢综合征,线粒体ROS增加,产生氧化应激,促进了miR-193b表达,导致肺动脉血管功能障碍,而miR-193b表达增加又导致核因子Y α亚基(nuclear factor Y α subunit,NFYA)表达下降,进而促使可溶性鸟苷酸环化酶β1(soluble guanylate cyclase,SGCβ1)合成减少,环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)降低,最终导致肥胖心衰大鼠出现运动诱导的肺动脉功能障碍[32]。在血管紧张素II(angiotensin II,Ang II)诱导的小鼠心肌肥大模型中,miR-106a表达增加,并靶向抑制线粒体融合蛋白2(mitofusin 2,Mfn2)表达,造成线粒体膜电位去极化,线粒体嵴缺损,ROS增多,心肌肥大;而抑制miR-106a或过表达Mfn2可改善上述现象,缓解心肌肥大[33]。miR-181c由核基因组编码后转位到线粒体发挥调控作用,其表达增加可靶向抑制线粒体呼吸链复合体I(mitochondrial respiratory chain complex I,mt-COX I)的表达,导致mt-COX IV结构重塑和功能障碍,造成线粒体ROS和基质Ca2+浓度增加,线粒体功能受损,线粒体代谢改变[34]。而敲除miR-181c可减少线粒体ROS产生,减轻氧化应激,降低糖酵解水平,改善线粒体功能,从而保护缺血性心衰小鼠心肌功能。过表达miR-181c还可通过下调mt-COX I的表达减少线粒体ROS产生,进而降低特异性蛋白1(specificity protein 1,Sp1)的表达,使线粒体钙摄取蛋白1(mitochondrial calcium uptake 1,MICU1)的表达下降,实现对线粒体基质钙水平调控;而敲除miR-181c可通过上述通路促进MICU1表达,降低线粒体钙摄取,减轻小鼠缺血再灌注损伤[35]。在心衰大鼠中,miR-665表达升高,并靶向抑制胰高血糖素样肽1受体(glucagonlike peptide 1 receptor,GLP1R)表达,降低mt-COX I、mt-COX III、mt-COX IV及超氧化物歧化酶(superoxide dismutase,SOD)活性,造成氧化应激和线粒体损伤,脂质过氧化产物丙二醛增多,并表现出心肌肌丝溶解,肌膜受损,线粒体数量减少,且大小不一、空泡化、肿胀等结构变化;而抑制miR-665可上调GLP1R表达,提高线粒体呼吸链复合体活性,降低氧化应激,同时还可减轻炎症反应,激活cAMP信号通路,减少心肌细胞凋亡的发生,改善心肌纤维和线粒体超微结构和心脏收缩功能[36]。在糖尿病患者和糖尿病模型小鼠血清中,miR-340-5p表达均上调,过表达miR-340-5p可导致糖尿病小鼠心肌纤维化、心肌肥大,并靶向抑制髓细胞白血病基因1(myeloid cell leukemia-1,MCL-1)的表达,促进ROS增加,SOD活性下降,造成氧化应激,同时,mt-COX I和mt-COX IV活性降低,使线粒体呼吸能力和ATP合成水平降低,并导致线粒体嵴断裂、空泡化、密度降低,进而影响线粒体功能,促进心肌细胞凋亡,而抑制miR-340-5p可通过促进MCL-1表达改善上述病理变化[37]。
心脏所需能量主要来自线粒体氧化代谢,在心衰过程中,心肌代谢由脂肪酸氧化为主转为糖酵解为主,葡萄糖成为主要的能源底物,在这一能量供应模式转变过程中,线粒体受到异常表达的miRNA调控出现功能异常[9,38]。研究显示,敲除miR-181a/b可通过上调胞质磷酸酶张力蛋白同源蛋白(phosphatase and tensin homolog,PTEN),抑制PI3K-Akt信号通路,造成小鼠心脏收缩功能下降[39]。在心衰小鼠模型中,心脏miR-146a表达增加,miR-146a可靶向抑制位于线粒体基质中的二氢硫辛酸琥珀酰转移酶(dihydrolipoyl succinyl transferase,DLST),影响线粒体氧化代谢,造成心肌肥大和心脏功能障碍;而敲除miR-146a或过表达DLST有助于维持线粒体氧化代谢水平,保护心脏功能[40]。去乙酰化酶3(sirtuin 3,SIRT3)可通过乙酰化调控激活线粒体代谢通路的多种关键酶,如调节脂肪酸氧化的长链酰基辅酶A脱氢酶(long-chain acyl coenzyme A dehydrogenase,LCAD)、调节三羧酸循环的琥珀酸脱氢酶(succinate dehydrogenase,SDH)、异柠檬酸脱氢酶2(isocitrate dehydrogenase 2,IDH2)等,进而调控多种病理生理变化[41-43]。在心衰患者或动物模型中,miR-195表达增加,其可结合线粒体SIRT3的3'UTR,抑制其表达。细胞实验显示,过表达miR-195可降低SIRT3水平,同时使丙酮酸脱氢酶复合体和ATP合酶乙酰化水平增加,导致线粒体呼吸功能下降,ATP合成减少。在小鼠心肌中特异性过表达miR-195也会出现同样的现象,且小鼠表现出心肌肥大和心衰症状[44]。这表明miR-195可通过调节SIRT3的表达影响蛋白乙酰化水平进而影响心肌功能。SIRT3缺失还会促进Ang II诱导的心肌肥大的发展,并最终形成原发性心衰。过表达miR-214可抑制SIRT3的表达,并可通过SIRT3/FoxO3a信号通路降低Mfn2的表达,并使Drp1表达下降,造成线粒体呼吸功能受损,线粒体出现肿胀、嵴断裂、空泡化等形态变化;抑制miR-214则可改善线粒体呼吸功能,改善心肌肥大[45]。miR-142-3p则在压力负荷诱导的大鼠心肌肥大模型中表达下降,并使src同源物2B接头蛋白1(src homology 2(SH2) B adaptor protein 1,SH2B1)表达增加,造成心肌肥大、线粒体膜电位和呼吸功能下降,线粒体密度降低;过表达miR-142-3p可抑制SH2B1表达,提高线粒体膜电位、呼吸功能和线粒体密度,改善线粒体功能,减少心肌细胞凋亡,缓解心肌肥大进程,预防心衰[46]。
线粒体蛋白稳态平衡是维持线粒体正常功能的关键因素。研究表明,慢性心衰患者miR-574表达增加,并可通过靶向调节位于线粒体的跨膜蛋白——序列相似性家族210成员A(family with sequence similarity 210 member A,FAM210A)的表达,影响线粒体编码的蛋白质表达,特别是线粒体呼吸链复合体的表达,进而调控线粒体蛋白稳态和线粒体功能,影响心肌肥大和心功能[47]。实验证实:过表达miR-574可提高FAM210A表达,进而导致线粒体编码蛋白表达增加,但不影响核基因编码蛋白;而敲除miR-574可降低FAM210A表达,造成线粒体编码的蛋白质表达下降,使线粒体膜电位受损、ATP合成减少,mt-COX I、mt-COX III、mt-COX IV活性降低,且伴随有严重的线粒体肿胀、嵴紊乱,进而促进心肌肥大、凋亡和心肌重塑。miR-1a-3p过表达可通过促进线粒体DNA编码的NADH脱氢酶1(NADH dehydrogenase 1,ND1)和细胞色素氧化酶1(cytochrome c oxidase I,COX I)的表达,改善ISO诱导的心衰小鼠的心肌结构与功能障碍[48]。糖尿病心肌病早期多伴随有心脏早期舒张功能不全、左室重构、心肌肥大、心肌纤维化等症状,并最终可能发展为心衰。miR-762是一种由核基因编码的转位到线粒体发挥调控作用的miRNA,其在缺氧刺激后表达上调,抑制NADH脱氢酶亚基2(NADH dehydrogenase subunit 2,ND2)的表达,影响线粒体功能,降低mt-COX I活性和氧化磷酸化水平,促进ROS产生,进而促进心肌细胞凋亡,导致心梗发生;而敲低miR-762或过表达ND2可提高线粒体ATP含量和mt-COX I活性,减少细胞凋亡,改善心肌功能[49]。在心衰患者或动物模型中,心肌miR-152表达增加,研究显示,心肌特异性过表达miR-152可通过靶向抑制线粒体谷氧还蛋白5(glutaredoxin 5,Glrx5)表达,影响线粒体铁硫蛋白稳定性,造成铁超载,导致线粒体发生基质电子密度降低、嵴断裂、外膜组织紊乱、线粒体尺寸变小为特征的形态异常、随机分布,同时伴有琥珀酸脱氢酶亚基表达下降,进而造成心脏收缩功能下降,纤维化程度增加,而抑制其表达可改善压力负荷诱导的心衰小鼠心肌纤维化和收缩功能障碍,保护线粒体结构[50]。
综上,miRNA在心衰发展中发挥了重要的线粒体调控作用。而且miRNA分布十分广泛,除在胞质中外,越来越多位于线粒体、内质网等细胞器中的miRNA相继被发现。由于功能和结构差异等因素,miRNA在不同位点的分布和表达量也不尽相同。对于线粒体相关的miRNA,根据其来源和定位可以分为3类:靶向线粒体转录物的核基因组编码的胞质miRNA、核基因组编码的线粒体miRNA、线粒体基因组编码的线粒体miRNA,后两者均定位于线粒体,统称为线粒体miRNA(mitochondrial miRNA,mitomiR)[51]。但目前对线粒体基因组编码的miRNA研究相对较少,如前所述,大多为核基因组编码miRNA。而核基因组编码的miRNA是如何进入线粒体的,目前尚不十分清楚,推测有以下几种可能的机制[21]:一是由于mRNA/miRNA相互作用能或细胞应激引起的热力学不稳定性导致GW182从RNA诱导沉默复合体(RNA-induced silencing complex,RISC)中裂解,游离的AGO2(argonaute 2)蛋白和来自RISC的miRNA通过线粒体外膜孔蛋白SAM50或TOM20进入线粒体内膜,而后在线粒体内膜蛋白的作用下,如TIM等,进入线粒体基质,至于AGO2蛋白和miRNA是组合进入还是分开进入的,目前尚不清楚;二是在磷酸化修饰作用下,如AGO2蛋白磷酸化,miRNA与mRNA结合被抑制,促进了AGO2/miRNA复合体形成,激活该复合体与P小体合并,P小体可以与线粒体、内质网等细胞器作用,使miRNA进入线粒体内;近来也有研究报道,AGO2/miRNA复合体可能通过多核苷酸磷酸化酶(polynucleotide phosphorylase,PNPase)的帮助进入线粒体外膜和内膜。miRNA 3'端的附加序列UGUCGGUGAGU也可能诱导了miRNA向线粒体的转位,如miR-181a和miR-181c均有此序列[52]。
2.2 lncRNA对心衰进程中线粒体功能的调控作用
lncRNA是一种长度大于200个核苷酸的非编码RNA,其可通过调节基因转录、翻译、翻译后修饰、表观修饰、蛋白质或RNA稳定性等调控细胞周期、细胞分化、细胞代谢等多种生理病理过程[53]。lncRNA可作为miRNA的分子海绵发挥调控作用,也可直接绑定目的蛋白甚至编码小肽。最近研究显示,lncRNA还可直接与信号分子相互作用[54]。由于lncRNA自身即是功能单位,因此lncRNA的亚细胞定位对于其功能发挥尤为重要[53]。lncRNA虽然主要定位在细胞核,在细胞质以及线粒体和内质网等细胞器中也广泛存在。研究显示,ncRNA占到人线粒体转录组的15%,其中,lncRNA在线粒体基因调控中更是发挥了关键作用[53]。lncRNA在调控线粒体功能影响心衰过程中同样发挥了多样的调节作用(表2)。

Table 2 Regulation effect of lncRNA on mitochondrial function and its mechanism during heart failure表2 lncRNA对线粒体功能的调控及影响心衰进程的机制
lncRNA可通过调控靶miRNA或蛋白质调控线粒体氧化应激,影响心衰进展。研究表明,lncRNA-Plscr4是一种内源性分子海绵,对miR-214具有负调控作用,过表达Plscr4可降低miR-214表达,进而促进Mfn2表达,减少ROS产生,改善线粒体膜去极化,保护线粒体功能,减轻Ang II诱导的心肌肥大[55]。MitoQ是一种线粒体ROS抑制剂,可有效降低氧化应激,改善线粒体功能。研究显示,MitoQ可以促进Plscr4和Mfn2表达,降低线粒体ROS和去极化水平,减轻氧化应激,并提高心衰小鼠线粒体氧化代谢相关蛋白质的表达,改善心衰小鼠线粒体和心肌功能[56]。lncRNA-DACH1是一种高度保守的lncRNA,主要位于细胞质中,DACH1可通过靶向调节SIRT3影响线粒体氧化应激与细胞凋亡进而调控糖尿病心肌病(diabetic cardiomyopathy,DCM)小鼠心脏功能[57]。在DCM小鼠和高糖处理的心肌细胞中,过表达DACH1可促进SIRT3泛素化降解,导致线粒体ROS增多、膜电位和MnSOD活性下降、ATP合成减少,线粒体变短和碎片化,进而促进细胞凋亡;而敲除或敲低DACH1可提高SIRT3蛋白水平,降低氧化应激,提高线粒体膜电位和抗氧化水平,改善线粒体结构与功能损伤,减少心肌细胞凋亡。在线粒体来源的lncRNA中,lncRNA细胞色素b(cytochrome b,cytb)是由线粒体DNA编码的复合体III的亚基转录而来,在胞质和线粒体中均表达[58]。细胞和动物实验均显示,抑制cytb会促进氧化应激和细胞凋亡,加重心肌损伤,而过表达cytb可提高线粒体功能,减轻心肌损伤,改善心衰。研究显示,过表达cytb可靶向抑制miR-103-3p的表达,进而导致miR-103-3p靶蛋白PTEN表达增加,缓解心肌肥大,减少线粒体ROS,改善心衰,而敲低cytb则导致ROS和细胞凋亡增加,促进心肌肥大。lncRNA-RMRP与热休克蛋白70(heat shock protein 70,HSP70)均在炎症调控中发挥关键作用,研究显示,脂多糖诱导的脓毒症小鼠心肌RMRP表达下降,线粒体膜电位降低,膜通透性增加,ROS产生和细胞色素c释放增加,促进了心肌细胞凋亡,过表达RMRP可通过靶向抑制miR-1-5p,促进HSP70蛋白4(heat shock protein 70 protein 4,HSPA4)的表达,减少ROS产生,改善心肌线粒体损伤,降低心肌细胞凋亡[59]。
心肌细胞代谢状态的改变是心梗、心肌肥大等病理性心脏变化的重要特征,也是促进心衰发展的重要因素。研究显示,一种在心肌组织中富集的lncRNA-lncHrt,可能作为一种心肌代谢调控因子,通过调节心肌代谢影响心脏功能[60]。在心梗发生后lncHrt表达明显下降,对周期蛋白依赖性激酶5(cyclin-dependent kinase,CDK5)抑制作用减弱,导致其与SIRT2相互作用加强,从而抑制胎肝激酶B1(liver kinase B1,LKB1)对腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)的磷酸化作用,造成线粒体氧化磷酸化和脂肪酸代谢水平下降,最终造成心肌重塑、心脏功能下降;而过表达lncHrt可改善上述情况,并通过抑制CDK5与SIRT2作用,促进LBK1磷酸化,激活LBK1-AMPK信号通路,增强线粒体氧化磷酸化水平,保护心脏功能。lncRNA-Caren是一种主要位于胞质中的lncRNA。Caren缺失会促进压力负荷诱导的心衰小鼠心脏收缩功能下降、心肌肥大和心衰发展,增加小鼠死亡率,同时,伴随心衰发生,组氨酸三聚体核苷结合蛋白1(histidine triad nucleotidebinding protein 1,Hint1)表达增加,ATM-DDR(ataxia telangiectasia mutated-DNA damage response)通路被激活,造成线粒体膜电位和呼吸功能下降,进一步促进线粒体和心脏功能障碍;过表达Caren可提高心衰小鼠心肌线粒体DNA和呼吸链复合体蛋白表达水平,促进线粒体生物合成与氧化磷酸化,并通过抑制Hint1表达抑制ATMDDR通路,保护心脏功能,减轻心肌肥大和纤维化水平,缓解心衰[61]。
线粒体相关的自噬与凋亡,参与线粒体质量控制,和神经退行性病变、心衰等疾病密切相关,在胚胎发育、细胞分化和炎症反应等的调控中也发挥重要作用[62]。在ISO诱导的心梗模型中,lncRNAMALAT1在梗塞组织中表达增加,并可绑定其靶miRNA-miR-558,抑制其表达,进而使miR-558的靶基因Unc-51样自噬激活激酶1(unc-51 like autophagy activating kinase 1,ULK1)表达增加,细胞凋亡水平下降,促使细胞保护性自噬增加;细胞实验证实过表达MALAT1可提高线粒体膜电位、减少ROS产生,降低细胞凋亡水平[63]。这表明,MALAT1可通过线粒体依赖性MALAT1-miR-558-ULK1通路调节细胞凋亡和自噬,改善心梗和心脏功能。作为急性心梗标志物以及内源性的肌质网Ca2+-ATPase 2a(sarcoplasmic reticulum Ca2+-ATPase 2a,SERCA2a)的抑制剂,lncRNA-ZFAS1表达增加可造成心梗小鼠细胞钙超载和心肌收缩功能障碍[64]。研究显示:敲低ZFAS1可改善缺氧诱导的线粒体肿胀和嵴断裂,提高线粒体膜电位和细胞活性,降低心梗和缺氧条件下线粒体介导的细胞凋亡;而过表达ZFAS1可通过抑制SERCA2a,造成细胞钙超载、线粒体膜电位降低、线粒体肿胀,进而激活线粒体介导的细胞凋亡通路,促进细胞凋亡。DACH1也可直接绑定SERCA2a。在小鼠心肌细胞中特异性过表达DACH1可促进SERCA2a泛素化降解,降低其蛋白质水平,使钙瞬变减少,心脏收缩功能障碍;而敲除DACH1可提高SERCA2a蛋白水平和钙瞬变,降低细胞凋亡,缓解心肌肥大,改善心衰小鼠心脏功能[65]。在缺血性心衰大鼠和缺氧刺激的H9C2心肌细胞中,lncRNASOX2-OT表达升高,实验证实,SOX2-OT可作为miR-215-5p的分子海绵抑制其表达,进而影响后者靶蛋白E盒结合锌指蛋白2(zinc finger E-box binding homeobox 2,ZEB2)的表达。过表达SOX2-OT可促进细胞凋亡和胶原蛋白堆积,而敲低SOX2-OT可抑制miR-215-5p,促进ZEB2蛋白表达,降低细胞凋亡,减轻心肌纤维结构紊乱和炎性细胞浸润,改善心衰,但敲低SOX2-OT未能改善心肌细胞线粒体功能障碍[66]。
lncRNA与miRNA之间也并非只是单向调控关系。Xu等[67]证实,lncRNA-CASC7与miR-30c相互调控影响心衰进程。在心衰患者血清中CASC7显著升高,而miR-30c显著下降。在细胞实验中,过表达CASC7可抑制miR-30c表达,同时促进白介素-11(interleukin-11,IL-11)表达,而miR-30c对CASC7和IL-11均表现为抑制效果,因此,二者均有可能是心衰诊断的标志物。尽管二者调控心衰的具体机制并未明确,但研究显示,miR-30和miR-133可抑制结缔组织生长因子,二者表达下降也会促进衰竭心肌组织的纤维化[68]。另外,IL-11也具有促进心肌细胞纤维化的作用[69],因此,CASC7和miR-30可能通过调控心肌纤维化影响心衰发展。
lncRNA具有很强的组织特异性,广泛的选择性剪切特性也使lncRNA具有多种定位模式,进而使其功能更加多样化[53]。lncRNA的不同定位是由其序列决定的,特定基序的存在或缺失促使其与特定的RNA结合蛋白或染色质相互作用,进而引导其输出到胞质或保留在核内[53]。与miRNA一样,lncRNA也可由核基因组或线粒体基因组编码,且两种lncRNA可以互相转运,并在协调细胞内区室间交流中发挥重要作用[70]。但lncRNA在细胞核与线粒体之间的转运机制并不清楚,有推测认为,核基因组编码的lncRNA转位到线粒体可能与线粒体膜的跨膜通道蛋白相关,也可能是通过特定的囊泡系统传递的;而线粒体基因组编码的lncRNA是如何转运到细胞核的尚未可知[70]。
2.3 circRNA对心衰进程中线粒体功能的调控作用
circRNA是一种单链环状RNA,大部分circRNA是由前体mRNA的外显子区域、内含子区域、外显子-内含子区域或编码蛋白质的基因的tRNA的内含子区域反向剪切形成,剪切后的RNA的3'端和5'端以磷酸二酯键相连,形成环形RNA[71]。circRNA功能广泛:调控基因转录,调节剪切;作为蛋白质支架或修饰基因调节亲本基因表达;作为蛋白质海绵调节蛋白质表达;作为miRNA海绵调节miRNA;形成circRNP复合物调节信号通路;作为竞争性内源性RNA与mRNA相互作用调节其转录;结合蛋白质影响mRNA翻译;多肽或蛋白质翻译等[72]。与线性RNA不同,circRNA没有位于5'端的7-甲基鸟苷帽子结构,也没有位于3'端的多聚赖氨酸尾。而且,由于circRNA的闭合环状结构特点,其相比miRNA和lncRNA更具稳定性,半衰期也更长,使其在细胞外液中含量丰富进而更容易被检测到,因此,circRNA可能作为一种良好的心血管疾病生物标志物[71]。尽管近年来关于circRNA调控各种病理生理变化的研究逐渐增多,但关于circRNA调节线粒体功能影响心衰作用的研究还很少(表3)。
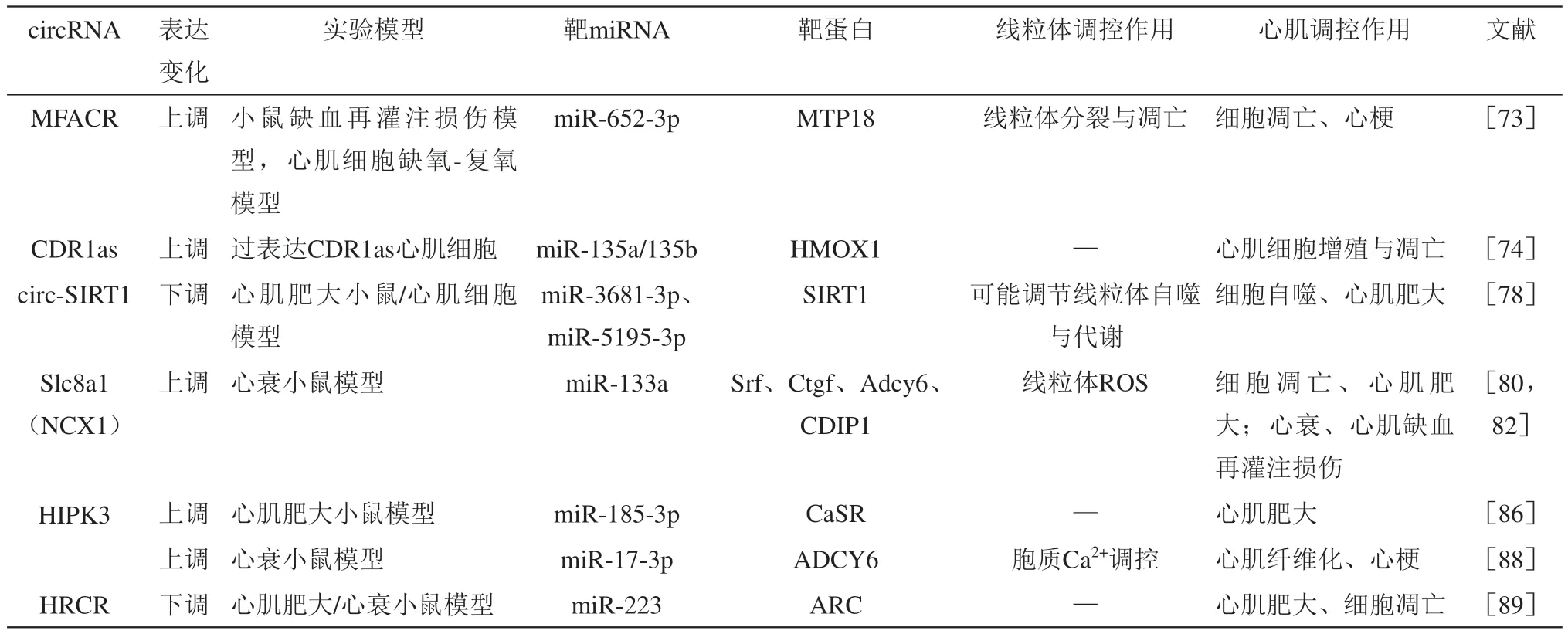
Table 3 Regulation effect of circRNA on mitochondrial function and its mechanism during heart failure表3 circRNA对线粒体功能的调控及影响心衰进程的机制
研究显示,circRNA-MFACR可通过靶向调控miR-652-3p影响线粒体分裂过程蛋白(mitochondrial fission process 18 kDa protein,MTP18)表达,调节心肌线粒体分裂与细胞凋亡进而调控心梗或缺血性心肌损伤[73]。MFACR可作为miR-652-3p的分子海绵抑制其表达,导致后者靶蛋白MTP18表达增加,诱导线粒体分裂加强和细胞凋亡;敲低MFACR可促进miR-652-3p表达,降低MTP18蛋白水平,降低线粒体分裂和细胞凋亡水平。
CDR1as是一种参与多种癌症和神经疾病发生发展的circRNA,在慢性心衰患者血浆中CDR1as和血红素加氧酶1(heme oxygenase 1,HMOX1)表达均增加,miR-135a和miR-135b表达则下降。研究证实,CDR1as是miR-135a和miR-135b的分子海绵,可抑制其表达,进而通过影响HMOX1表达调控心肌细胞凋亡[74]。HMOX1是调节血红素代谢的关键酶,HMOX1及其代谢物被证实具有扩张血管、调节细胞增殖、抗氧化应激、抗炎症、抗凋亡、调节铁死亡等作用,在调控线粒体功能与质量控制中也发挥重要作用[75-76]。另外,Mester-Tonczar等[77]在猪缺血性心衰模型中检测到CDR1as在心梗组织中高表达,且CDR1as水平与心脏收缩功能改善呈正相关、与心梗面积呈负相关关系,蟾蜍灵处理可提高心梗组织CDR1as的表达,降低miR-671-5p水平,改善心脏收缩功能。因此,CDR1as可能作为慢性心衰诊断和治疗的有效标志物,并可能通过miR-135a/b-HMOX1轴调控线粒体功能,改善心衰发展。
去乙酰化酶1(sirtuin 1,SIRT1)的同源circRNA-circ-SIRT1可以通过竞争性吸附miR-3681-3p和miR-5195-3p,上调其宿主基因SIRT1表达,同时可以通过招募泛素特异肽酶22(ubiquitin specific peptidase 22,USP22)诱导SIRT1蛋白去泛素化进而在翻译后水平提高SIRT1蛋白稳定性,促进细胞自噬,缓解心肌肥大[78]。SIRT1可通过PGC-1α调节线粒体生物合成与代谢,并在线粒体自噬调控中发挥重要作用[79]。由此,circ-SIRT1可能通过miR-3681-3p、miR-5195-3p影响PGC-1α表达,进而调控线粒体功能。
源自钠钙交换器(sodium-calcium exchanger,NCX)基因第2外显子的circRNA-Slc8a1可以作为miR-133a的分子海绵调控压力负荷诱导的心肌细胞肥大[80]。抑制或过表达Slc8a1可降低或升高miR-133a靶蛋白血清应答因子(serum response factor,SRF)、结缔组织生长因子(connective tissue growth factor,CTGF)、β肾上腺素能受体1(adrenoceptor beta 1,ADRB1)和腺苷酸环化酶6(adenylate cyclase 6,ADCY6),进而缓解或促进心衰。而由NCX转录产生的另一circRNA-NCX1也可作为miR-133a的分子海绵,且在ROS刺激下表达增加,并通过调节miR-133a促进心肌死亡诱导蛋白(cell death-inducing protein,CDIP1)表达,促进心肌细胞凋亡,而抑制NCX1可通过miR-133a-CDIP1轴降低CDIP1表达,减少心肌细胞凋亡,缓解心肌缺血再灌注损伤[81]。miR-133a是调控心肌肥大的关键因子,其过表达可有效抑制心肌肥大[82],且miR-133a在调控线粒体形态、生物合成、复合体活性、呼吸功能、氧化磷酸化中具有重要作用[83-85]。因此,Slc8a1/NCX1-miR-133a-线粒体轴可能是调控多种心脏疾病的有效通路。
circRNA-HIPK3在肥大心肌细胞中高表达,研究证实,过表达HIPK3可靶向抑制miR-185-3p的表达,进而使其靶蛋白钙敏感受体(calciumsensing receptor,CaSR)表达增加,促进压力负荷诱导的心肌肥大和心脏功能障碍,敲除HIPK3可通过miR-185-3p-CaSR轴缓解压力负荷诱导的心肌肥大[86]。CaSR是一种G蛋白偶联受体,在维持细胞钙平衡、调节细胞增殖和凋亡中发挥重要作用,研究显示,CaSR激活可改善高糖诱导的线粒体结构与功能损伤,如线粒体分裂增加,长度变短,线粒体呼吸链复合体活性下降,ATP合成减少等[87]。HIPK3还可通过靶向调控miR-17-3p影响ADCY6的表达,调节胞质Ca2+浓度,敲低HIPK3可通过维持心肌细胞的Ca2+处理能力缓解心梗后心衰小鼠心肌纤维化,改善心脏功能[88]。心脏相关circRNA(heart-related circRNA,HRCR)是miR-223的分子海绵,可通过调节miR-223的表达,影响其靶蛋白-细胞骨架活性调节蛋白(activity-regulated cytoskeleton-associated protein,ARC)的表达,调控心肌肥大和心衰进程[89]。
尽管circRNA调控线粒体影响心衰的研究还较少,但综上可知,其调控作用不可忽视。特别是对于线粒体基因组编码的circRNA研究尚处于起步阶段,其详细机制有待深入探究。新近研究表明,线粒体基因组编码的circRNA在线粒体、细胞质和细胞外均有广泛分布,并且可以通过调控线粒体生物合成、氧化应激、蛋白质表达等影响慢性淋巴细胞白血病、非酒精性脂肪肝等多种疾病的发展,展现了线粒体来源circRNA潜在的多样调控能力[90]。此外,circRNA具有明显的细胞类型、组织类型和不同发育阶段的表达特异性[4],探究其在不同病理生理条件和阶段的差异化表达,对更好地认识ncRNA调控心衰等疾病的发生发展大有裨益。
综上表明,ncRNA可通过调控线粒体代谢、蛋白质稳态、融合与分裂、氧化应激、凋亡与自噬等多种生理活动,调节线粒体结构与功能变化,影响心肌损伤和心脏功能,调节心肌凋亡、肥大、心梗等心脏病理变化,进而影响心衰形成与发展(图1)。其中lncRNA和circRNA可通过ncRNA-miRNA-mRNA调控网络形式实现对线粒体结构与功能的调控,从而影响心衰的发生发展。同一种ncRNA可通过不同通路调节心脏病理变化,不同ncRNA也可能通过调节同一靶标调节线粒体与心脏结构与功能。
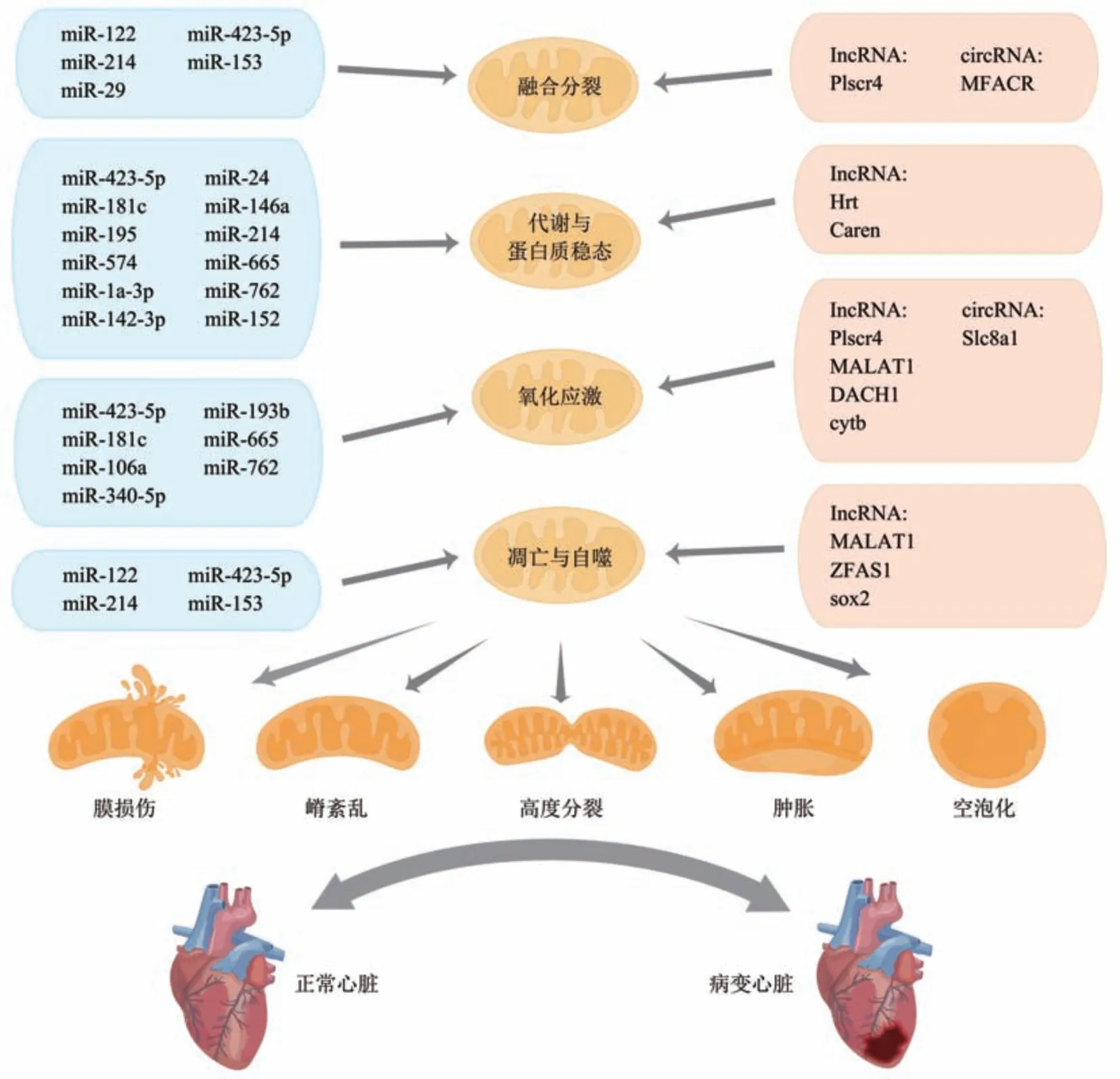
Fig. 1 The schematic graph of ncRNA regulating mitochondrial function during heart failure图1 非编码RNA调控线粒体功能影响心衰示意图
3 总结
心脏疾病发展过程中,ncRNA的异常表达被认为是机体的适应性调控,大量研究结果也证实ncRNA在心衰发展中具有重要的功能性调控作用,ncRNA-miRNA-mRNA调控网络研究也为心衰治疗提供了更多新的治疗靶点和思路[71,91]。线粒体在能量代谢、生物合成、信号转导、细胞死亡等生理病理过程调控中具有重要作用,线粒体功能障碍介导的细胞死亡也是心衰发展的重要因素[1]。近年来,线粒体基因组来源的ncRNA的发现也进一步证明,线粒体ncRNA在各种疾病调控过程中发挥了不可替代的重要作用。除线粒体基因编码的miRNA、lncRNA、circRNA外,线粒体基因组编码的多种长度小于200个核苷酸的ncRNA,如piRNA和由核糖体RNA、转运RNA、lncRNA等衍生出的小片段ncRNA,也具有明显的组织特异性,并被证实在多种疾病调控中发挥了重要作用[90]。由此,从生物发生、表达模式、代谢特点、调控作用等方面对线粒体基因组编码的ncRNA的研究或许将开启一个新的领域[90]。围绕线粒体定位的ncRNA,探究ncRNA-mRNA网络对线粒体功能的调控机制及其在心脏疾病中的作用,可以为心脏疾病诊断和治疗提供更多理论依据和新的治疗靶点,为药物研发提供新的作用靶标。可以期待在未来临床治疗中,通过纳米颗粒包装等新技术手段,将线粒体ncRNA靶向注射到患者心脏中,实现早期干预治疗,延缓发病甚至治愈心脏病变。不过,关于ncRNA对其靶基因或靶蛋白的调控作用仍有待进一步阐明。核基因组和线粒体基因组来源的ncRNA在不同细胞器之间的定位、转运机制及其对不同细胞器甚至细胞间交流的协调作用也有待进一步明确。这些更深层次问题的解决,将有助于更好地认识ncRNA在调控线粒体进而影响疾病发展中的角色,为临床研究与治疗提供更丰富的选择手段。
