7例Gilbert综合征临床分析及文献复习
2022-10-28古再丽努尔吾甫尔李雪国毛敏
古再丽努尔·吾甫尔,李雪国,毛敏
(新疆维吾尔自治区人民医院血液病科,新疆 乌鲁木齐830000)
0 引言
Gilbert综合征(GS)是一种最常见的胆红素葡萄糖醛酸化遗传性疾病。临床症状轻微,可表现为轻度、非结合型高胆红素血症、波动性黄疸,肝脏一般无器质性改变。非结合型高胆红素血症的最常见病因包括胆红素生成过多、GS以及新生儿黄疸。GS临床需要与溶血性贫血等血液病鉴别,临床容易误诊或漏诊,多数预后良好,不需特殊治疗[1]。因此我们对2018年2月至2021年10月期间我院收治确诊的7例GS临床资料进行回顾性分析,现报道如下。
1 临床资料
1.1 一般资料
2018年1月至2021年3月在我科住院确诊的7例Gilbert综合征;男性5例,女性2例,最小年龄22岁,最大年龄49岁;汉族3例,维吾尔族2例,回族2例,病史最长30年,最短1年。见表1。
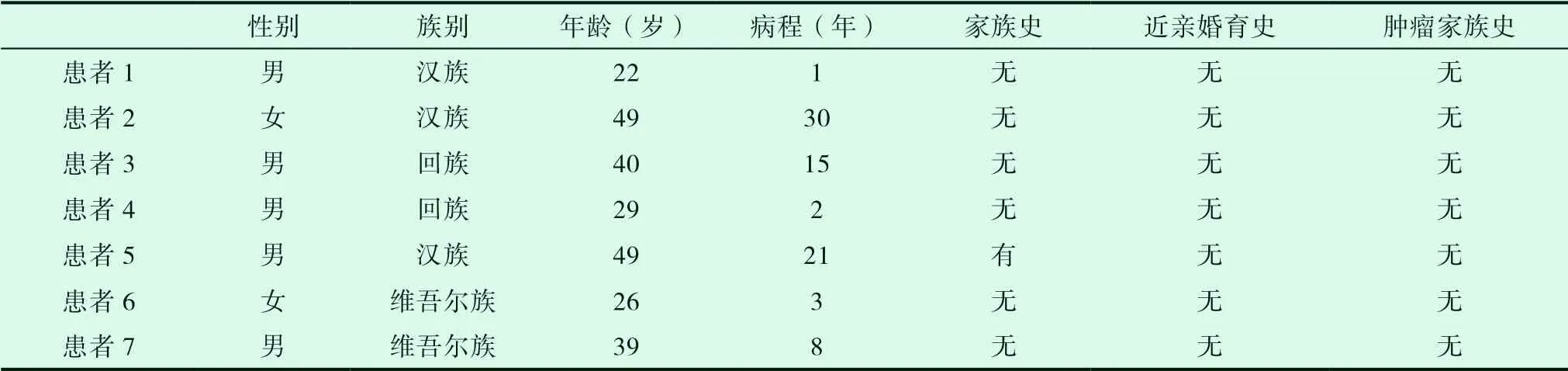
表1 患者一般情况
1.2 观察指标
患者一般情况观察指标一般临床资料、血常规、网织红细胞计数、肝功能、病毒学指标、自身免疫抗体、肝炎系列、肿瘤标记物、补体水平、免疫球蛋白、腹部B超,并均行骨髓检查。
2 结果
2.1 临床表现
7例患者均有皮肤巩膜黄染,乏力3例,食欲下降1例,恶心1例,上腹部饱胀不适1例。皮肤黏膜黄染诱发因素有:劳累4例,受凉2例,感染1例,饮酒1例。
2.2 辅助检查结果
7例均有总胆红素、间接胆红素轻、中度升高,其余肝功能指标均正常。甲、乙、丙肝病毒及CMV、EBV等病毒检查均阴性;ANA、ENA、dsDNA、SMA、AMA等抗体阴性; 2例患者网织红细胞计数增高,余检测指标均未见异常。7例骨髓涂片和骨髓病理未见异常。腹部B超检查7例患者肝脏、胆囊未见明显异常。见表2。
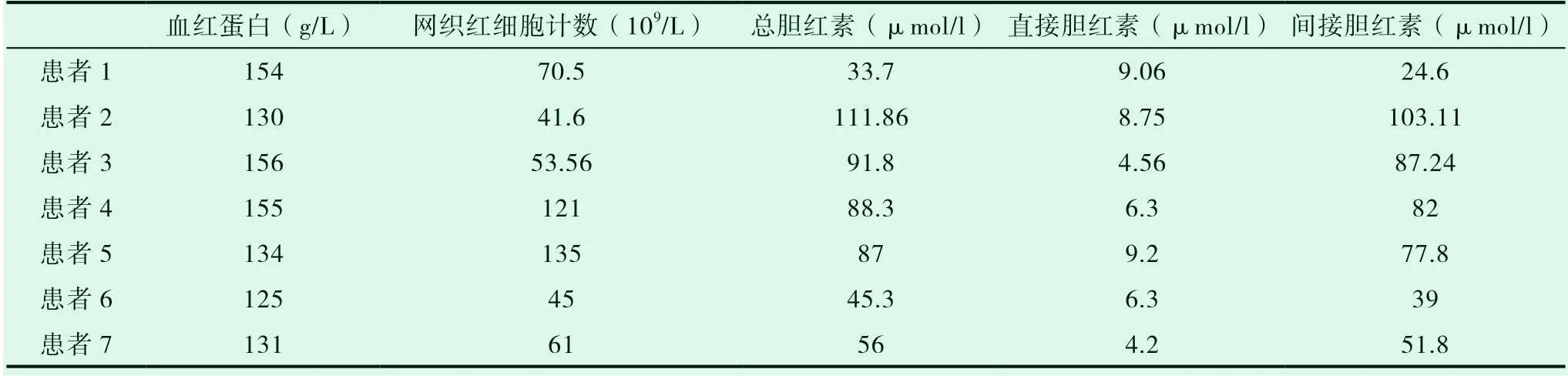
表2 血红蛋白、网织红细胞计数、胆红素水平
2.3 UGT1A1突变基因位点
7例患者均进行UGT1A1基因检测,本检测用PCR和基因测序的方法检测标本中UGT1A1的基因编码区1-5外显子以及基因上游苯巴比妥反应增强元件(PBREM)的突变分析,涵盖了该范围内的点突变、插入和缺失型突变。共进行5个PCR扩增反应和19个基因序列测定反应,7例患者均发生UGT1A1突变基因,见表3。
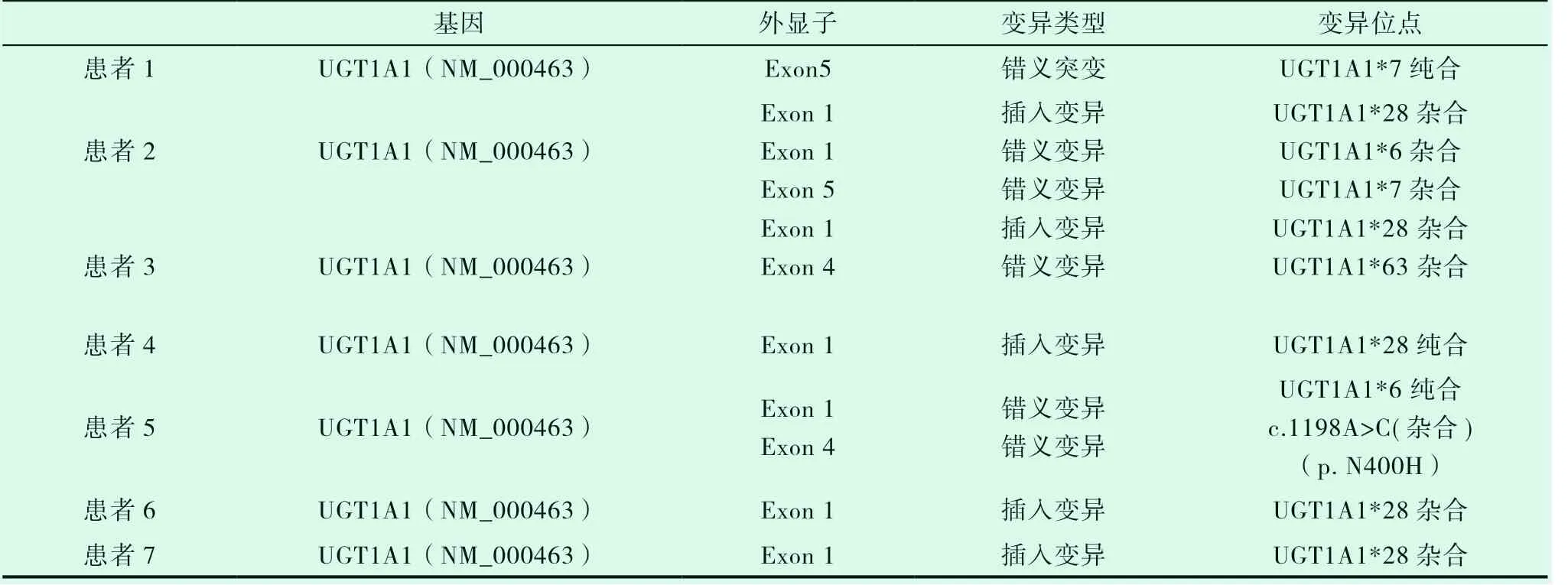
表3 UGT1A1突变基因位点检测结果
7例患者中4例为UGT1A1*28杂合突变,该位点为突变热点。患者4黄疸劳累、感冒等使黄疸加重,红细胞计数和血红蛋白正常,但红细胞形态小细胞低色素性,网织红细胞计数比例增高,骨髓涂片和活检提示红系增生明显活跃,溶血相关检查结果示FHb升高,Hp轻度减低,基因检查发现UGT1A1*28纯合突变,β地中海贫血基因CD14-15杂合突变,追问家族史发现父亲也有常年间歇性轻度巩膜黄染病史,父母脾脏不大,血常规、胆红素均正常,基因筛查发现患者父母均有UGT1A1*28纯合突变,父亲同时伴有β地中海贫血基因CD14-15杂合突变,但父母均未发病。患者5同时存在地中海贫血基因CD14-15杂合突变和UGT1A1基因突变,可确诊为β地中海贫血(轻型)和Gilbert 综合征同时存在。患者家系UGT1A1基因检查结果,见图1。
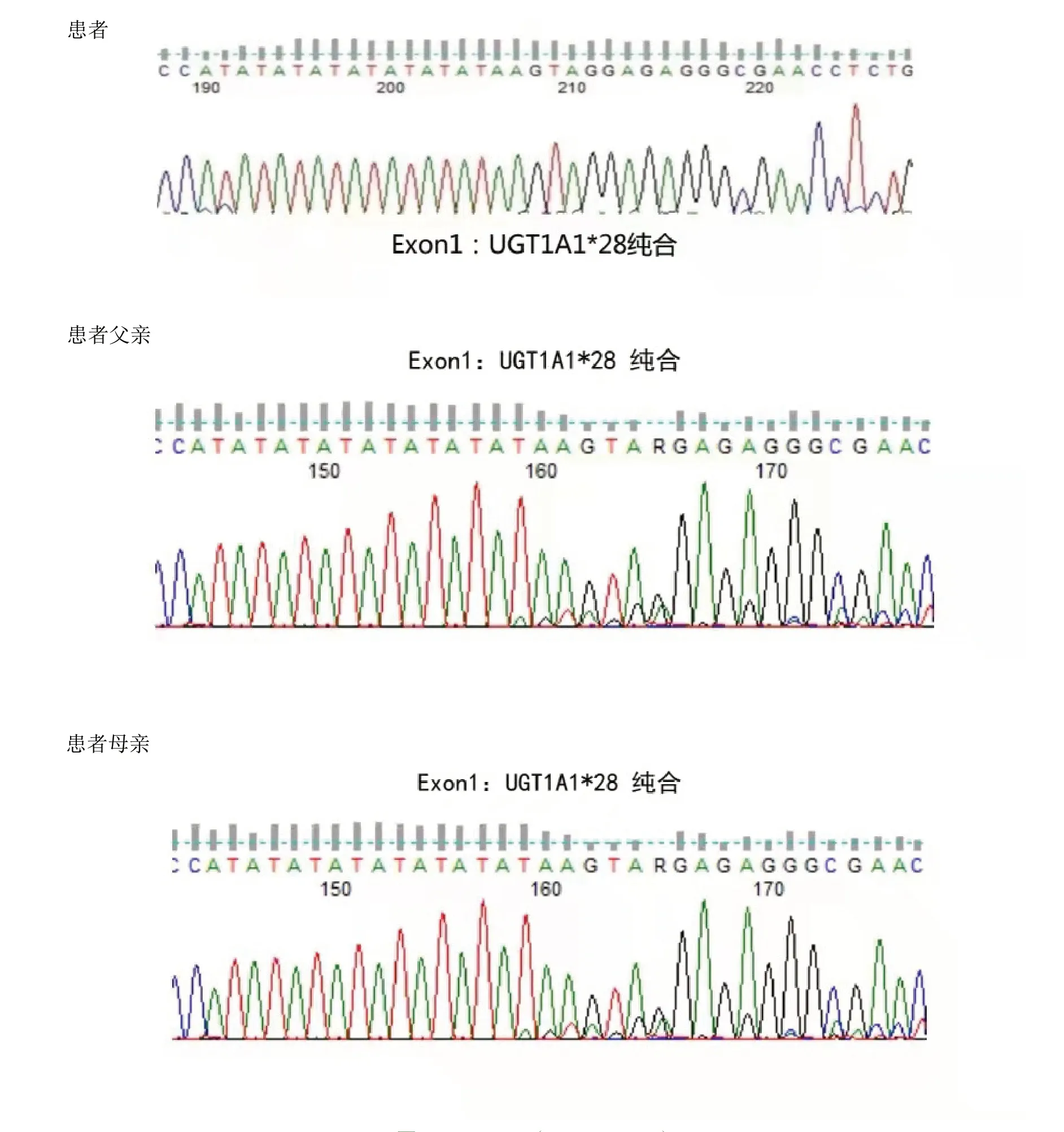
图1 UGT1A1(NM_000463)
3 讨论
Gilbert综合征由Gilbert于1901年第1次报道,是一种胆红素代谢障碍的常染色体隐性遗传性疾病。GS多在青春期前后或成年期被诊断。男性多见,多有家族史。UGT1A1基因突变导致尿苷二磷酸葡萄糖醛酸转移酶(UGT)蛋白表达水平下降,从而引起血清非结合胆红素水平升高,出现相应临床表现。纯合子往往发病较重,而杂合子病情较轻[2]。该文中患者4为UGT1A1*28纯合,患者5为UGT1A1*6纯合子突变,胆红素水平较其他患者明显增高,与相关文献相符。
GS的遗传机制与位于染色体2q37位点UGT1A1基因遗传多态性密切相关[3]。其中UGT1A1*28纯和突变多见。本文中7例患者中4例存在UGT1A1*28杂合突变,一例UGT1A1*28纯合突变,其余两例分别为UGT1A1*7纯合和UGT1A1*6纯合突变,UGT1A1*28杂合突变为热点突变。
尽管GS是一个良性过程,但Gilbert基因型被证实与胆石症风险显著相关[4],而在表型正常的人群则没有这一风险; 另外 GS 和遗传性球形红细胞增多的复合遗传也增加了胆结石的风险。患者4黄疸劳累、感冒等使黄疸加重,轻度小细胞低色素性贫血贫血,网织红细胞计数比例增高,黄疸以间接胆红素升高为主,骨髓涂片和活检提示红系增生明显活跃,检查结果示FHb升高,Hp轻度减低,基因检查发现UGT1A1*28错义突变,β地中海贫血基因CD14-15杂合突变,父亲也有间歇性巩膜黄染病史,父母脾脏不大,血常规、胆红素均正常,父母均有UGT1A1*28纯合突变,父亲也发现同时伴有地中海贫血基因CD14-15杂合突变,但均未发病。该患者同时存在地中海贫血基因CD14-15杂合突变和UGT1A1基因突变,可确诊为β地中海贫血(轻型)和Gilbert 综合征同时存在。国内李露锋[5]等报道过一例中间型ɑ-地中海贫血同时携带UGT1A1*28突变病例及其家系报告案例,先症者患者的父亲和女儿均缺失一个ɑ地贫基因,均为ɑ地贫,其父亲网织红细胞略高,女儿为UGT1A1*28携带者,但二者胆红素均正常,其母亲缺失了2个ɑ地贫基因,为轻型地贫。Lee HJ[6]等报告一例28岁男性患有胆结石和脾肿大,合并遗传性球形红细胞增多症(HS)和GS的患者。国内Jun Jiang[7]等报道过一例同时存在Dubin-Johnson syndrome和GS的突变的双重遗传性黄疸患者,国内仅报道1例。本文中患者4目前随访两年,血红蛋白正常,无合并症,一般情况,仍在随访当中。
Gilbert综合征患者一般不需特殊治疗,及时的诊断也有助于避免不必要的过度治疗。因UGT1A1酶活性降低,Gilbert综合征患者对某些药物的敏感性增强,易产生药物性肝损伤。国外研究认为UGT1A1*28能改善霍奇金淋巴瘤的预后、降低子宫内膜癌的风险[8]。本文中2例胆红素明显增高患者予以根据胆红素水平定期口服熊去氧胆酸胶囊降胆红素治疗后胆红素下降,余患者未予以特殊治疗,病情稳定。
综上,Gilbert综合征是临床上最为常见的一种先天性黄疸,对于胆红素增高、但无溶血患者需排查GS,UGT1A1基因测序检测是特异性及敏感度高的有效手段,可降低临床黄疸误诊误治,节约医学资源。对于高胆红素血症患者,全面筛查相关潜在因素非常必要,在临床表现无法用一元论解释的时候,也应该从多元论角度启动相关全面检查。
