基于ddGBS的黑杨派杨树SNP位点挖掘
2022-09-26刘粉粉姜小龙翁文源龚细娟徐刚标
刘粉粉,姜小龙,翁文源,龚细娟,徐刚标
(1.中南林业科技大学 林学院,湖南 长沙 410004;2.湖南省泰格林纸集团有限公司,湖南 岳阳 414000)
杨树Populus是杨柳科Salicaceae 杨属Populus的统称,染色体组为2n=38,生长迅速、适应性强、无性繁殖和有性繁殖容易,是北半球中纬度地区重要的工业用材林、防护林及园林绿化树种[1]。其中,黑杨派(SectionAigeiros)优良无性系在全球杨树人工林产业中占有重要地位[2],世界上90%的杨树优良无性系是从黑杨派的美洲黑杨P.deltoides和欧洲黑杨P.nigra种内或种间杂交子代中选育出来的。由于黑杨派无性系种质遗传基础狭窄,亲缘关系较近,很难鉴别形态特征差异[3]。
分子标记揭示的DNA 多态性,是现代遗传学研究的基础,也是物种分类、种质鉴定的强有力工具[4]。Rajora 等[5]采用SSR 和RAPD 标记鉴定欧美杨无性系,Chauhan 等[6]利用AFLP 标记鉴别缘毛杨P.ciliata与马氏杨P.maximowiczii杂交子代,宋红竹等[7]和李善文等[8]分别采用AFLP标记分析杨树无性系间、种间、派间及亲本与子代间亲缘关系,郭丽琴等[9]采用SRAP 标记分别研究杨属不同种亲缘关系,贾会霞等[10]、郑涛等[11]及刘超凡等[12],先后采用SSR 标记构建杨树无性系指纹图谱。但是,这些标记类型都有其局限性,不同学者使用的分子标记类型及其引物不同,可能会导致研究结果不能共享。
单核苷酸多态性(single nucleotide polymorphism,SNP)是单碱基突变引起的DNA 序列多态性。虽然单个SNP 位点的多态信息含量较低,但数以百万计潜在的全基因组SNP 位点或小插入/缺失的信息,为高通量基因分型奠定了基础[4,13]。SNP标记摒弃了凝胶电泳检测技术,位点多为双等位、共显性,基因分型结果便于记录和分析解读,适合自动化分析,在不同的检测技术间重现度高[13-14]。但是,挖掘海量SNP 位点的高通量基因测序分型,耗时且十分昂贵[4]。采用酶切技术针对物种基因组特定区域进行简化基因组测序,降低了测序量和测序成本,已成为挖掘SNP 位点信息的主要方法[4,15]。
2011年,Elshire 等提出的基因分型测序(Genotyping by sequencing,GBS),相对于其它简化基因组测序技术,简化了文库的构建过程,不受参考基因组限制,无需进行片段大小选择,加快了DNA 序列数据获取的进度,尤其对多态性低、重复序列高的物种,具有独特的优势[16],已被广泛应用于SNP 标记开发、物种分类、种质鉴定、遗传多样性分析、基因组选择、遗传图谱构建及分子辅助选择育种[4,14,18-20]。杨树SNP 标记研究报道较多[18-20],测序方法多为从头测序(De novo sequencing)[18]、酶切简化基因组测序RADseq (Restriction-site associated DNA sequencing)[19]和转录组测序[20-21],基于GBS 技术挖掘杨树基因组SNP 位点信息,鲜有报道[22]。本研究以46 份欧美杨无性系种质为材料,利用ddGBS 技术挖掘高质量SNP 位点信息,旨在为杨树SNP 标记开发、指纹图谱构建、遗传多样性分析以及重要经济性状关联分析等奠定前期研究基础。
1 材料与方法
1.1 材 料
46 份黑杨派无性系样本采自湖南省岳阳市泰格林纸集团有限责任公司国家杨树种质资源库。采集的叶片样品,用硅胶快速干燥后,保存在-4℃冰柜中。
1.2 方 法
1.2.1 DNA 提取
采用天根试剂盒提取基因组DNA,0.8%琼脂糖凝胶电泳测定DNA 质量,核酸蛋白仪测定DNA 的浓度与纯度。
1.2.2 GBS 测序文库构建
采用MseI(T|TAA)和TaqaI(T|CGA)双酶切基因组,T4DNA 连接酶连接其两端的普通接头和条形码(barcode)接头,全自动磁珠纯化系统回收纯化连接后的片段。以回收片段为模板,进行PCR扩增,混合样本,再次回收纯化。利用 Illumina Hiseq2000 高通量测序平台,进行2×145 bp双末端测序。GBS 文库的构建及测序,委托上海美吉生物医药科技有限公司完成。
1.2.3 测序数据质控
采用Fastp v 0.19.6(https:// https://github.com/OpenGene/fastp)软件,对原始数据进行质控。质控标准为:去除双端测序序列中接头序列和含N比例大于10%的序列,5′端质量评分低于Q20(碱基错误识别概率为1%)、3′端质量评分低于Q30(错误识别概率为0.1%)的碱基,以及超过40%碱基质量评分低于Q15(合格)的序列。
采用FastQC v 0.11.9(https://github.com/s-andrews/FastQC/archive/ v0.11.9.tar.gz)软件,评估质控后的数据。利 用Stacks v2.55(http://catchenlab.life.illinois.edu/stacks/)软件中的process-radtags 程序,参数选择默认值,将质控后的序列片段截成长度为140 bp,用于后续SNP 变异位点开发。
1.2.4 SNP 位点开发
采用Bowtie2 v2.3.2(http://bowtie-bio.sourceforge.net/index.shtml)软件,将剪切后的数据比对到毛果杨(P.trichocarpa)参考基因组(https://www.ncbi.nlm.nih.gov/assembly/GCF_0000027 75.4)上。采用Samtools v1.12(http://www.htslib.org/) 软件,按照SNP 位点在基因组上的位置进行排序。运行Stacks v2.55 软件中的Stacks-gstacks 程序,将测序片断组装为完整、无断点的重叠克隆群,再比对到位点上,识别每个SNP 位点;运行Stackspopulations 程序,对检测到的 SNP 位点数据进行过滤。SNP 位点过滤标准为:次要基因型频率(Minor allele frequency,MAF)>0.05,完整性>0.8,最大杂合度为1,最小测序深度为 6,无缺失数据。
1.2.5 SNP 位点注释与染色体定位
利用SnpEff v4.3(http://pcingola.github.io/SnpEff/)软件,对SNP 位点进行注释。运行R 语言中的“CMplot”包,绘制SNP 位点在染色体上的分布图;运行R 语言中的“ggplot2”包,统计SNP 位点数目,分析位点数目与染色体长度的相关性并绘制相关性曲线图。
2 结果与分析
2.1 测序质量
46 份黑杨派无性系种质经ddGBS 测序后,得到的原始图像数据经碱基识别分析转化的原始测序序列数据量为108 Gb,每个样本平均数据量为2.35 Gb。去除低质量的数据后,获得的高质量数据的基本信息见表1。共产生218 156 221 条序列片断,总读长数据为62 832 509 890 bp;样本序列片段数目为2 795 062(NL1383)~7 994 127(ZKY),平均片断数目为4 742 526条。各样本读长数目在802 475 700(NL1383)~2 295 941 115 bp(BKY) 范围内,平均读长为 365 924 128 bp。Q30 测序质量值为93.32%~94.44%,平均值为93.87%。所有样本的Q30 值均在93%以上,说明碱基测序错误率较低,获得的测序数据合格。GC 含量为41.83%(Y706)~44.96%(Z13),平均值为42.65%。GC 含量普遍不高,分布正常,完全符合测序的要求。
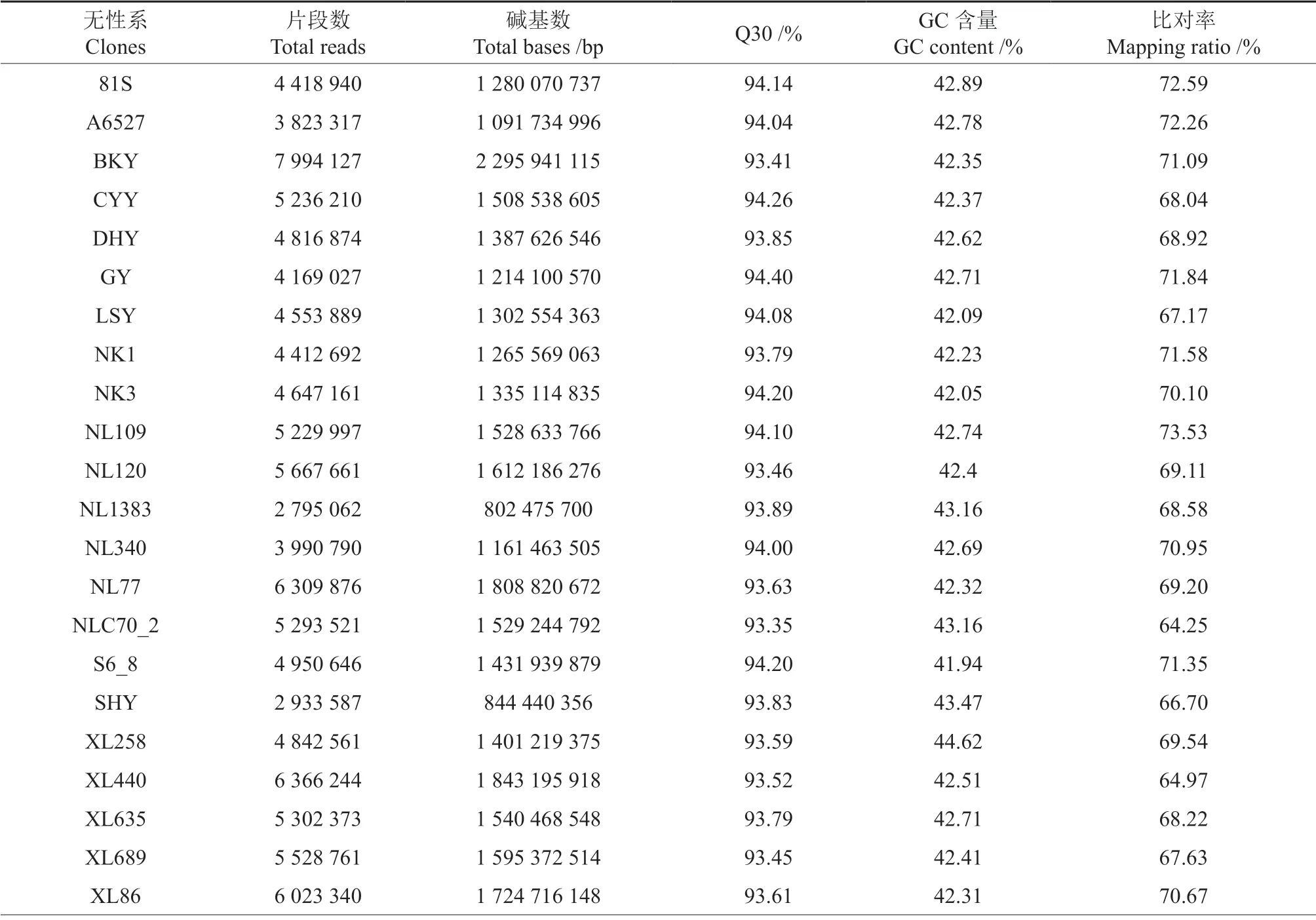
表1 杨树GBS 测序数据Table 1 GBS sequencing data of 46 Populus

续表1Continuation of table 1
FastQC 软件对质控后数据进行评估的结果,见图1。图1中的前145 bp 和后145 bp 分别代表测序片段两端的质量值分布情况。由图1A 可知,碱基G、C 含量波动范围为18%~25%;从图1B可以看出,片断碱基测序的平均错误率低于0.04%。采用Bowtie2 软件,将所有样本序列比对到毛果杨基因组上,比对率为64.25%(NLC70_2)~73.53%(NL120),平均为69.89%(表1),比对率较高,这表明参试样本基因组与毛果杨参考基因组有较高的相似性。位点测序深度为12.4(Z7)~24.7(81S),平均值为18.4。
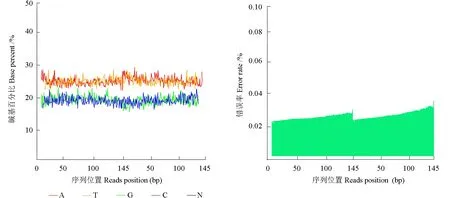
图1 双端碱基含量及测序质量值Fig.1 Distribution and sequence qualities of double-terminal bases
2.2 SNP 变异类型及其在基因组中的分布特征
46 个黑杨派无性系种质材料中,共鉴定出386 558 个SNP 变异位点。运行Stacks-populations程序,获得131 194 个特异性SNP 位点。SnpEff软件对SNP 位点注释的结果,见图2A。6 种可能的碱基突变中,C/T(38 056)和A/G(37 694)突变最多,分别占总突变数量的29.00%和28.73%。转换与颠换的比例为1.37∶1,其中,同义突变19 543 个,错义突变20 368 个,无义突变225 个。
分析基因组中SNP位点分布的结果,见图2B。SNP 变异位点主要位于基因下游、上游和转录区,分别占基因组总SNPs 数量的29.48%、25.51%和19.06%;分布于外显子、内含子和基因间隔区的SNPs 占比分别为8.09%、8.10%和5.95%;3′和5′非翻译区(Untranslated region,UTR)分布的SNPs数目较少(3.38%),位于剪切体上的SNP 位点更少(0.43%)。
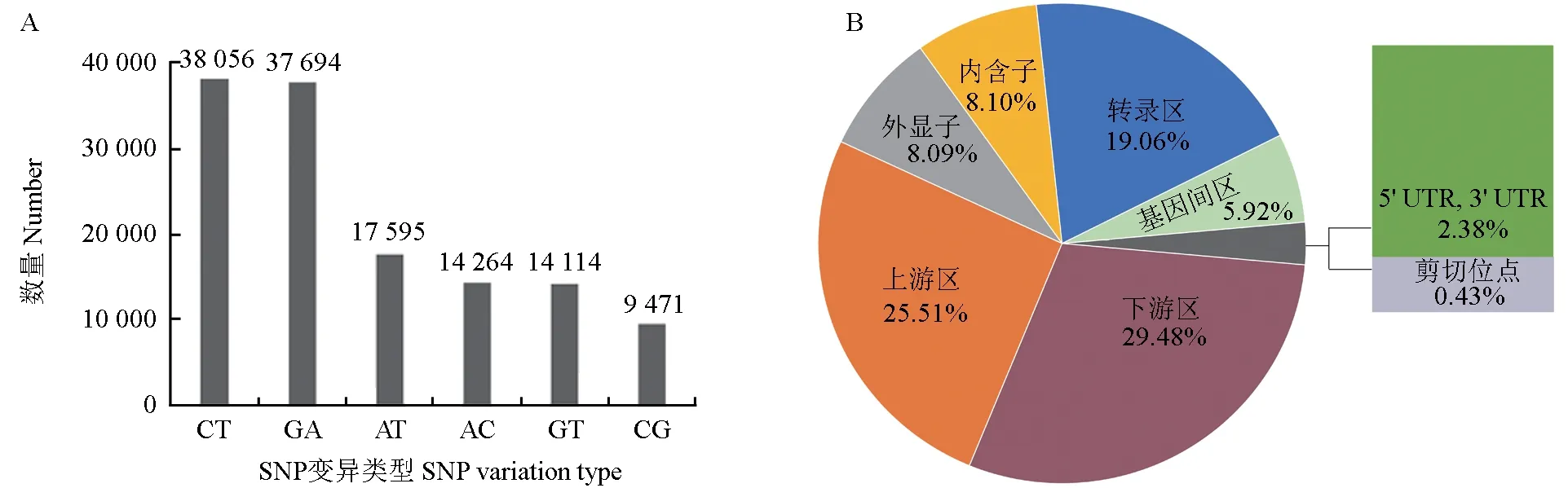
图2 SNPs 注释结果图Fig 2 The annotation of SNPs in the Populus genome
2.3 SNP 位点在染色体上分布特征
运行R 语言中的“CMplot”包,绘制的染色体上SNP 位点分布图(图3A)表明,98.41%(129 104)的SNP 位点能成功定位到19 条染色体上,每条染色体分布的SNP 位点数目为3 402(染色体19)~15 566(染色体1),平均分布密度为1/3 188 bp。染色体长度与SNP 数目相关性分析的结果显示(图3B),染色体长度与SNP 数目呈极显著线性正相关(r=0.84,p=1.97e-08)。这表明,SNP 位点在染色体上分布均匀。

图3 染色体上SNP 位点分布情况分析Fig.3 Distribution of SNP loci on the chromosomes
3 讨 论
限制性内切酶的酶切是各种简化基因组测序的共同点和起点[4,14-16]。通过对酶切片段测序,降低基因组的复杂度。因此,选择合适的限制性内切酶是构建高质量文库的关键。不同物种,选择的限制性内切酶种类及其组合不同,其酶切效率存在差异[23]。五碱基酶Ape KⅠ(G|CWGC)是Ⅱ型限制性内切酶,DNA 转座子区几乎没有识别位点,且部分甲基化敏感,常用于简化基因组测序。四碱基酶因其酶切的片段最小,也是常用的内切酶。采用ApeKI 单酶切美洲山杨(P.tremuloides)基因组[22],四碱基限制性内切酶Hae Ⅲ(GG|CC)单酶切金花茶(Camellia nitidssima)[24]和古茶树(Camellia sinensis)[25]基因组,效果较为理想。与单酶切相比,双酶切更大程度上降低了基因组复杂度,增加了构建文库的一致性,基因组上的位点分布更加均匀[14]。采用六碱基酶PstI(CTGCA|G)和四碱基酶MspI(C|CGG)组合,开发出较理想的桉树(Eucalypts)基因组SNP 位点[26]。本研究采用四碱基酶MseI 和TaqaI 双酶切杨树基因组,构建的文库质量较高。
本研究基于ddGBS 技术对46 份欧美杨无性系种质进行测序,数据过滤后,共获得131 194个SNPs 变异位点,SNPs 分布密度为1/3 308 bp,接近于美洲山杨(160 183)[22]和拟南芥Arabidopsis thaliana(1 /3300 bp)[27]基因组中SNPs 分布密度,远低玉米Zea mays(1/120 bp~1/60 bp)[28]、水稻Oryza sativa(1 /268 bp)[29]、大豆Glycine max(1/185~1/260 bp)[30]和黄麻Corchorus capsularis(1/172 bp)[31]基因组中的SNPs 分布密度。这可能与物种基因组结构与大小的差异有关。杨树基因组相对较小(434 Mb,https://www.ncbi.nlm.nih.gov/genome/?term=Populus.+trichocarpa),大部分基因组序列属于编码序列,存在于非编码序列中的SNP 会提高SNP 出现的频率[32]。也可能与检测样本大小及测序长度有关[33]。本研究中,挖掘的潜在SNP 变异位点数目(386 558)仅为毛果杨转录组(561 302)[18]的3/5,这是由于ddGBS 技术获得的测序片段数相对较少,对基因组覆盖率低[14]。
不同物种间转换与颠换的比值存在很大差异,大多数DNA 片段中,转换发生频率比颠换发生频率要高[34]。甜瓜Cucumis melo基于2b-RAD 测序的结果,转换与颠换比率为2.15[35];GBS 技术检测雪茄烟草Nicotiana tabacum基因组SNP 位点发现,转换与颠换比率为1.24[36];青杨Populus cathayana转录组测序的结果,转换与颠换比率为1.57[21]。本研究中,转换与颠换比率为1.37。与产生这种差异的原因,可能与不同物种在其长期进化过程中承受的进化选择压力不同有关[37]。转换类型中,C/T 突变最多(29.00%),这可能与CG序列上SNP 变异位点出现的最为频繁,胞嘧啶(Cytosine,C)常以甲基化形式存在,脱氨后成为胸腺嘧啶(Thymine,T)有关[38]。
注释后的杨树基因组中的SNPs,主要位于基因上下游和转录区,部分位于基因间区和内含子区域上,8.09%的SNP位点位于外显子功能区域上,说明部分变异位点对蛋白翻译有影响,这可能是杨树基因组相对较小、大部分基因组序列属于编码序列。本研究中,出现在基因间区和内含子上的SNP 位点数目相接近,这与基于转录组测序的结果基本一致[18],98.41%(129 104)的SNP 位点能成功定位到19 条染色体上,表明研究的材料与毛果杨亲缘关系很近。SNPs 数目与染色体长度呈极显著的线性正相关,说明这些SNP 位点在染色体上分布均匀。
GBS 技术最显著的优点是,DNA 用量少,建库效率高效,成本低廉,发掘的SNP 位点在基因组中分布均匀,比其他各种基于高通量测序平台的基因分型技术具有成本竞争力,克服2b-RAD片段过短,易受重复干扰的不足,会使育种工作者在不必开发任何标记的前提下进行物种分类、种质鉴定、基因组选择或分子辅助选择育种[14]。但是,ddGBS 获得的测序片段数较少,对基因组覆盖度相对降低。在今后的研究中,将杨树转录组数据与ddGBS 测序数据相结合,将会提高SNP位点的分布密度和基因定位效率。
4 结 论
本研究表明,采用ddGBS 能有效地挖掘杨树高质量SNP 位点信息。ddGBS 具有高通量、低成本等特点,今后可用GBS 技术建立涵盖杨树国家种质资源库所有种质材料的简化基因组基因分型数据,构建较为完整的种质无性系DNA 指纹图谱,为杨树无性系种质鉴定和新品种知识产权保护提供理论依据,并为进一步开展杨树种质遗传多样性分析及重要经济性状关联分析,乃至杨树分子辅助选择育种提供了重要的遗传信息。
