UPLC-MS-MS 检测猪肉中的甲硝唑原药及其代谢产物羟基甲硝唑
2022-08-09张光前朱贵平
黄 霞,张光前,朱贵平
(宜宾市叙州区食品检验检测中心,四川宜宾 644699)
畜禽养殖中常用各类抗生素作为畜禽疾病常用药,有些药物间隔期还未到期就上市销售,长久食用会产生抗药性,危害消费者的身体健康。硝基咪唑类抗生素是畜禽饲料中常用的抗原虫感染及抗菌药,具有促生长作用,可与其他抗生素联合用于治疗厌氧菌感染[1-6]。
目前测定硝基咪唑类药物的方法主要有气相色谱法、高效液相色谱法、气相色谱串联质谱法和高效液相色谱串联质谱法[7-10]等。其中色谱检测因为定型能力弱,易检出假阳性。而气相色谱串联质谱法适用面窄,且其处理复杂。高效液相色谱串联质谱法选择特征离子扫描,抗干扰能力强,且检出灵敏度高,具有检出使用范围广等优点,广泛应用于各类兽药残留的检测中。
1 材料与方法
1.1 试剂与仪器
甲硝唑标准品(中国药品生物制品检定所);羟基甲硝唑标准品(WITEGA 99.4%);二氯甲烷、乙酸乙酯、乙腈、甲醇、甲酸及丙酮均为色谱纯(山东大禹);氯化钠为分析纯(成都科隆);实验用水为超纯水(舒活泉)。
AB4500Q 液质联用仪(配有电喷雾离子源和大气压化学源 美国AB 公司);CP225D 电子天平(赛多利斯);LD4-2C 离心机(北京京立离心机有限公司)。
1.2 实验方法
1.2.1 对照品溶液的配制
精密称取待测组分甲硝唑及其代谢产物羟基甲硝唑标准品各10.0 mg,甲醇溶解为1 000 μg/mL 标准储备液。分别精密吸取甲硝唑、羟基甲硝唑标准储备液0.010 mL 于同一个100 mL 容量瓶中,甲醇定容至刻度,得到1 000 ng/mL 混合标准中间液。分别精密吸取0.010 mL、0.020 mL、0.050 mL、0.100 mL和0.500 mL 于进样瓶中,阴性样品提取液定容至1.00 mL,得到浓度为10 ng/mL、20 ng/mL、50 ng/mL、100 ng/mL 和500 ng/mL 混合标准工作液。
1.2.2 样品前处理方法
准确称取均匀样品约10.0 g 于50 mL 离心管中,加入5 g 硅藻土与样品充分混匀后在加入2.5 mL 饱和氯化钠水溶液,20 mL 甲醇+丙酮(3+1)。充分振荡混匀,超声30 min,离心,取上清液于蒸发皿中(重复提取一次)。将两次提取的上清液60 ℃电热板蒸干只剩水相,将水相全部转移至另一50 mL 离心管中,加入氯化钠饱和溶液5 mL,10 mL 乙酸乙酯,无水硫酸钠3 g,离心,取有机相于25 mL比色管中(重复提取一次),60 ℃氮吹至近干,用1.0 mL 甲醇溶解,上机。
1.2.3 色谱检测条件
色谱柱:C18(250 mm×4.6 mm,5 μm);进样体积:5 μL;流速:1.0 mL/min;流动相:0.1%甲酸/水(A)和甲醇(B);梯度洗脱程序:0 ~5.0 min,95%~5%A,5.1 ~10.0 min,5%A;10.1 ~15.0 min,95%A。
1.2.4 质谱检测条件
离子源:电喷雾离子源(ESI);采集模式:多 反 应 离 子 监 测(Multiple Reaction Monitoring,MRM);扫描模式:正离子扫描;离子源温度:600 ℃;电喷雾电压:5 500 V;气帘气压力:35 psi;喷雾气压力:60 psi;辅助气压力:70 psi;其余质谱条件如表1 所示。
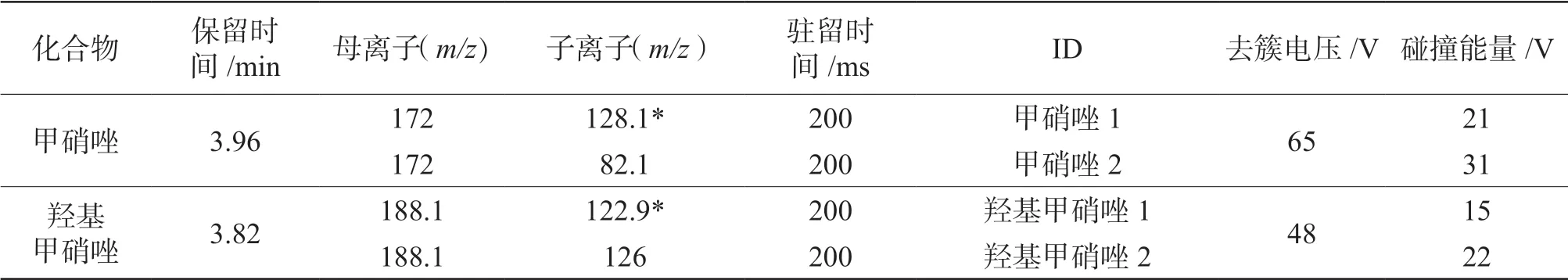
表1 待测组分质谱条件
2 结果与分析
2.1 样品前处理条件的优化
样品中加入硅藻土能更好地提取待测组分。硅藻土主要有3 个作用:粒间偶极作用;絮凝作用,能将待测组分形成絮凝物;吸附作用。这3 点都能促使待测组分更好地从动物组织中分离,吸附于硅藻土上。饱和食盐水则能更好地将动物组织分散开,使待测组分更易析出。
文献[6,8,10-12]中常用乙腈、酸性乙腈(含0.1%甲酸)、甲醇+丙酮(3+1)、二氯甲烷来作为萃取溶剂,提取动物组织中的兽药残留。本试验对这4 种提取溶剂的提取效果做了单因素实验。结果表明,当加标量为1 μg/kg 时,4 种萃取剂的回收率都较低,为40%~55%,当加标量为5 μg/kg、20 μg/kg 时,乙腈、酸性乙腈(含0.1%甲酸)回收率在45%~60%,回收率较低。二氯甲烷回收率在70%~80%。甲醇+丙酮(3+1)回收率在75%~85%,回收率最高。考虑到甲醇+丙酮(3+1)的毒性较乙腈和二氯甲烷低,且回收率较高,故选择甲醇+丙酮(3+1)为提取溶剂。提取溶液再经乙酸乙酯液液分配,能有效去除样品中的杂质,减少基质干扰。
文献中[6,8,10,12]还提到用凝胶渗透色谱,固相萃取小柱进行净化。凝胶渗透色谱溶剂用量大,仪器价格贵,未采用。采用C18固相萃取小柱净化,甲硝唑和羟基甲硝唑的回收率都较低,为50%~65%。在小柱净化的过程中,有一部分待测组分被C18颗粒吸附,未被洗脱,造成回收率偏低。
本实验采用液质联用的多反应离子监测模式进行采集,离子碎片的选择性高,能有效降低样品中的杂质干扰。因此,本实验通过乙酸乙酯液液萃取后,直接氮吹甲醇定容上机,能满足检测需要。
2.2 质谱条件的优化
将1.0 μg/mL 待测化合物的标准溶液在正离子模式下进行母离子全扫描,确定待测组分的母离子,再分别以待测化合物母离子,对其子离子进行全扫描,选择两个响应值较高,干扰较小的特征子离子作为子离子[11]。一般将响应值最高的作为定量离子,次之的作为定性离子(离子对见表1)。
将母离子的去簇电压(DP)设置为60 V,扫描5 ~180 V 的碰撞能量(CE),选择响应值最高的CE 值(具体值见表1),确定响应值最高时的CE 值后将DP 范围设置为5 ~300 V 扫描,得到响应值最高的DP 值(具体值见表1)。建立MRM 方法。
2.3 流动相的优化
流动相对化合物的出峰时间、峰形、响应值有直接影响。甲硝唑和羟基甲硝唑均为正离子采集模式,故采用含有0.1%甲酸的水溶液作为水相,酸性环境下,物质更容易电离成带正电荷的母离子。本实验比较了乙腈甲酸水与甲醇甲酸水的两种流动相对待测组分峰形的影响,实验表明,两者均有较好的峰形,且响应值差异较小。考虑到乙腈价格是甲醇的三倍以及乙腈毒性较甲醇更强,因此采用甲醇作为流动相。
2.4 线性范围、检出限及定量限
本实验以待测组分的浓度和色谱峰面积作为X、Y轴绘制线性回归方程。本实验标准曲线线性范围为10 ~500 ng/mL,甲硝唑、羟基甲硝唑的相关系数R2均大于0.999 9,通过向阴性猪肉样品中添加1 μg/kg待测组分,测定信噪比为3(S/N=3)时方法的检出限,信噪比为10(S/N=10)时方法的定量限。线性方程、线性相关系数、检出限及定量限,见表2,标准曲线见图1 和图2。

表2 回归方程、检出限、定量限
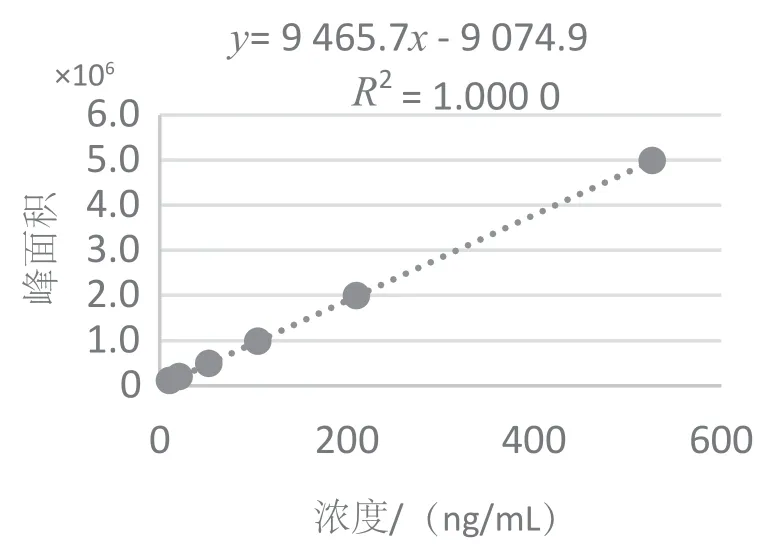
图1 甲硝唑定量离子标准曲线

图2 羟基甲硝唑定量离子标准曲线
2.5 方法回收率、精密度
选用阴性鲜猪肉样品,分别在1 μg/kg、5 μg/kg、20 μg/kg 低中高3 个加标水平对待测组分进行加标回收,高中低3 个浓度各测定6 次,结果见表3。5 μg/kg加标回收色谱图、阴性样品的色谱图见图3、图4。由此说明该方法的回收率基本满足检测需要。
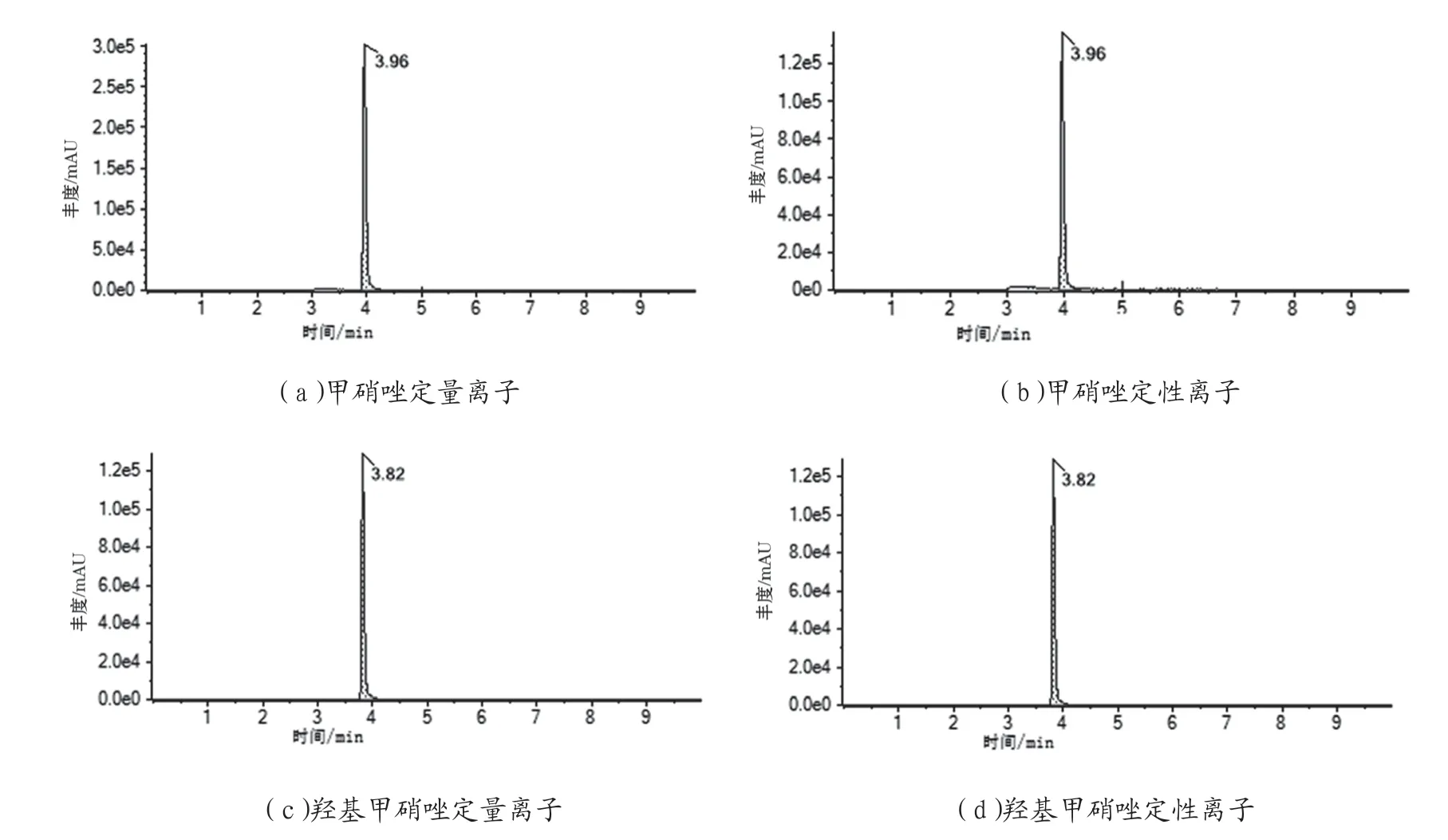
图3 5 μg/kg 加标回收色谱图

图4 阴性样品色谱图
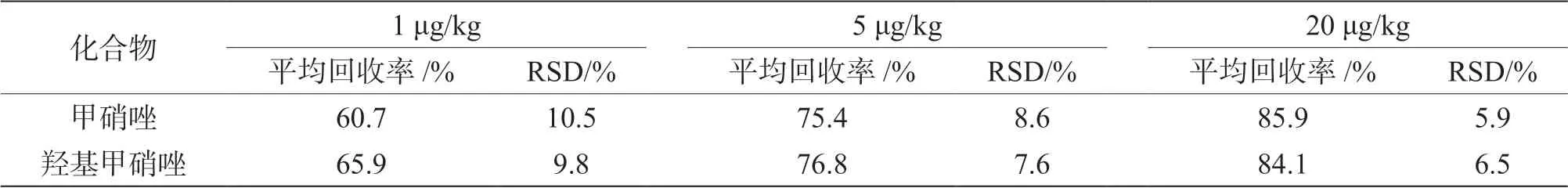
表3 回收率和相对标准偏差
2.6 实际样品分析
采用本方法对12 批鲜猪肉中的甲硝唑及羟基甲硝唑进行测定。每批样品从称样到检测完毕只需1 h。与传统固相萃取、凝胶渗透色谱法相比,样品分析时间缩减,且成本降低。实际检测结果显示,12 批鲜猪肉中的甲硝唑及羟基甲硝唑均未检出[13]。
3 结论
本实验通过对前处理和仪器检测条件的优化,基本满足检测需要。但是在含量较低时,回收率还有较大提高空间。如果能采用内标法定量,消除前处理中的损失及仪器检测时的基质效应,能有效提高方法的准确性,特别是样品含量较低时的定量准确性。
