病毒感染对NLRP3炎症小体活化、组装和效应的影响*
2022-07-25王志会
王志会 张 建
(山东大学药学院免疫药物学研究所,济南 250012)
2019 年12 月起,严重急性呼吸系统综合征冠状病毒2(SARS-CoⅤ-2)感染事件在全球大范围爆发,对全球经济造成巨大损失,公共健康与卫生安全受到严重威胁[1-2]。本次疫情将病毒重新拉回大众视野,无论是全球大流行的流感病毒(influenza virus)、人类免疫缺陷病毒还是在非洲肆虐的埃博拉病毒,都对人类社会产生了巨大危害。临床数据表明,有些病毒感染会引起细胞因子风暴,如ⅠL-6、TNFα、ⅠL-1β、ⅠFN-γ 等多种细胞因子迅速大量产生,这种现象是引起病毒性感染患者发生器官衰竭和呼吸窘迫综合征的重要原因。其中,ⅠL-1β 和ⅠL-18 通过增强吞噬细胞的抗菌特性,启动Th1 和Th17 的适应性免疫反应,促进宿主对感染的防御。与其他促炎细胞因子相比,ⅠL-1β 和ⅠL-18是由非活性前体pro-ⅠL-1β和 pro-ⅠL-18经过酶切割产生的具有生物活性的细胞因子,负责该加工处理的最重要的酶之一是胞内半胱氨酸蛋白酶caspase-1,该加工过程受到炎症小体的调控[3]。
炎症小体是由模式识别受体(pattern recognition receptor, PRR)、 接 头 蛋 白ASC(apoptosis-associated speck-like protein containing a CARD)和效应蛋白pro-caspase-1 组成的大分子多蛋白复合物,识别危险信号后,这些大分子复合物进行组装,触发caspase-1 的自我激活,进而对pro-ⅠL-1β 和pro-ⅠL-18 进行切割。同时,炎症型的caspase-1 可以切割gasdermin D(GSDMD),导致细胞焦亡[4-5]。炎症小体的活化有两个步骤:首先,宿主细胞对微生物的模式识别诱导了pro-ⅠL-1β 和NLRP3 的转录;其次,危险信号激活炎症小体,导致caspase-1 的活化,并将细胞因子前体切割为成熟的具有生物活性的ⅠL-1β和ⅠL-18,以及触发细胞焦亡[6-8]。炎症小体可分为依赖于caspase-1激活的经典型炎症小体和依赖于caspase-11/4/5 激活的非经典型炎症小体。目前已发现的能形成炎症小体的PRRs 包括NLRs(NOD-like receptors)和ALRs(AⅠM2-like receptors)家族蛋白等,其中对NLRP3炎症小体的研究最为广泛,在炎症反应和抗病毒反应中都发挥着重要作用。NLRP3 炎症小体激活和调控的机制复杂且存在争议。本文就NLRP3 炎症小体在病毒感染过程中的激活和调控以及病毒针对NLRP3 炎症小体的免疫逃逸机制等方面的研究进展作一综述。
1 病毒感染对NLRP3炎症小体活化的影响
静息细胞中基础表达水平的NLRP3 不足以形成炎症小体,需要细胞外炎症刺激来启动激活。例如,Toll 样 受 体(Toll-like receptors,TLRs)、NLRs 或细胞因子受体激活转录因子NF-κB,上调NLRP3 的表达水平并控制翻译后的修饰,来积累炎症小体的蛋白质组分。NLRP3 可被一系列不同的刺激物激活,这些物质包括晶体和微粒物质(如石棉、二氧化硅和明矾等)、细胞外ATP、成孔毒素、RNA-DNA混合物以及一些病毒、细菌、真菌和原生动物病原体。
1.1 第一信号
单独使用NLRP3激动剂不足以激活炎症小体,还需要一个启动信号,即第一信号。第一信号通常由微生物成分或内源性细胞因子提供,通过TLRs、ⅠL1R或TNFR等受体激活转录因子NF-κB。NF-κB是一个关键的炎症应答转录因子,通过上调NLRP3 和pro-ⅠL-1β 的表达提供组装炎症小体的蛋白质[9]。
不同的病毒感染对NLRP3 炎症小体的活化产生不同的作用。研究发现,乙肝病毒e 抗原(HBeAg)抑制脂多糖(lipopolysaccharide,LPS)引起NLRP3炎症小体的活化和ⅠL-1β的产生,通过晶体结构分析推测,HBeAg 通过与接头分子TRAM (TRⅠF-related adaptor molecule) 和MAL(MyD88 adaptor-like)结合,从而干扰NF-κB 信号通 路,抑 制NF-κB 磷 酸 化[10]。黏 液 瘤 病 毒(myxoma virus,MYXⅤ)的M013蛋白包含一个独特的33残基C端尾结构,可以直接与NF-κB结合,抑制炎症小体的活化[11]。严重急性呼吸道综合征冠 状 病 毒 (severe acute respiratory syndrome coronavirus,SARS-CoⅤ)ORF3a 蛋白,通过促进肿瘤坏死因子受体相关因子3 (TNF receptorassociated factor,TRAF3)依赖的p105 和ASC 泛素化,促进NF-κB 和NLRP3 炎症小体的活化[12]。在巨噬细胞中,手足口病病毒(foot-and mouth disease virus,FMDⅤ)RNA 诱导NF-κB 的活化,促进NLRP3和pro-ⅠL-1β的转录,激活NLRP3炎症小体和ⅠL-1β的分泌[13]。
1.2 第二信号
NLRP3 启动后可以被很多类型的化学和微生物刺激所激活,包括ATP、颗粒物质、细菌和病毒成分等。基于这些现象形成了一种假说,即NLRP3 并不直接识别这些刺激剂,而是通过检测细胞内的信号变化而发生活化。关于NLRP3 炎症小体激活的机制还存在激烈的争论,目前发现了几种常见的细胞内NLRP3上游信号,包括离子外流、线粒体损伤和ROS的产生、溶酶体破裂等。
1.2.1 离子外流
细胞的大部分能量用于维持细胞浆和细胞外环境之间的离子梯度。离子浓度的动态变化调控着细胞内许多酶和信号通路,在细胞信号传导中起关键作用,而且完整的离子梯度是防止健康细胞死亡的重要机制。NLRP3 激活剂可以诱导细胞产生离子浓度变化,包括K+外流、Ca2+动员等,这些都与激活NLRP3 炎症小体有关。越来越多的研究表明,某些RNA病毒编码的跨膜病毒孔蛋白(viroporins)通过形成膜通道改变膜对离子的渗透性,诱导细胞损伤或应激反应,RNA 病毒所导致的这些变化将激活NLRP3。
a.K+外流
K+外流被认为是NLRP3炎症小体的激活信号,几乎所有的NLRP3 刺激都会诱导K+外流,包括ATP、尼日利亚菌素和颗粒物质等,胞质K+浓度降低足以激活NLRP3炎症小体[14]。P2X7R是一个非选择性的阳离子通道,具有类似于K+通道的孔形成基序,因此被认为是一个K+外排通道,介导NLRP3 炎 症 小 体 的 激 活[15]。胞 外ATP 刺 激P2X7R, 触 发K+外 流, 引 起 泛 连 接 蛋 白1(pannexin-1)膜孔逐渐募集,P2X7 孔的打开也会使ATP 流出,从而放大活化信号。TWⅠK2 是一个K+外流通道,有研究认为它作为“刽子手”与P2X7R共同作用调节NLRP3炎症小体的激活[16]。
流感病毒M2 蛋白是一个H+选择性离子通道,当高尔基体腔呈酸性时,其定位于高尔基体膜上并引起H+外放,进而诱导NLRP3 炎症小体活化。另外,M2 蛋白的H37G 突变体诱导炎症小体活化的能力增加,这可能是因为突变体能在细胞膜上通过Na+/K+交换引起K+外流[17]。水泡性口炎病毒(vesicular stomatitis virus,ⅤSⅤ)或脑心肌炎病毒(encephalomyocarditis virus,EMCⅤ)的复制会触发K+外流,诱导NLRP3 炎症小体的激活,然后释放ⅠL-1β,导致细胞焦亡和坏死[18]。SARS-CoⅤORF3a蛋白直接或通过增强K+外排激活caspase-1,从而启动NLRP3炎症小体组装[19]。在马雅罗病毒(Mayaro virus,MAYⅤ)感染中,使用高浓度K+处理细胞会明显抑制ⅠL-1β 释放[20]。FMDⅤ2B 蛋白在细胞膜上形成孔道,诱导K+外排,作为第二信号激活NLRP3炎性小体。动物实验证实,2B蛋白增强机体针对FMDⅤ的特异性免疫反应,抑制病毒的复制,可作为自体佐剂增强口蹄疫病毒样疫苗或其他不需加强接种的亚单位蛋白疫苗的免疫应答[13]。
b.Ca2+动员
尽管Ca2+动员在许多信号通路中广泛存在,但是其在NLRP3 炎症小体激活中的作用仍然存在争议。在NLRP3 炎症小体激活过程中,干扰Ca2+信号会抑制炎症小体复合物组装、caspase-1 剪切和ⅠL-1β 分泌。细胞内Ca2+的增加如何促进NLRP3 炎症小体的激活尚不清楚[21]。一项研究表明,Ca2+的增加可以促进巨噬细胞中NLRP3 和ASC 之间的相互作用,直接调节NLRP3 炎症小体活化。丙肝病毒(hepatitis C virus,HCⅤ)感染仍然是导致肝脏炎症和疾病的一大诱因,其核心蛋白通过与磷脂酶c相关的Ca2+动员诱导炎症小体的活化,被认为是激活NLRP3炎症小体的关键因子[22]。据报道,小核糖核酸病毒(picornavirus)病毒孔蛋白2B 蛋白可以降低内质网和高尔基体中的Ca2+水平,导致Ca2+进入细胞质并增加局部Ca2+浓度,细胞质离子浓度失调导致NLRP3 炎症小体的活化[23]。同样,EMCⅤ和人类鼻病毒(human rhinovirus,HRⅤ)病毒孔蛋白2B蛋白特异性降低内质网/高尔基体的Ca2+浓度,并诱导NLRP3分布到核周区域,从而激活NLRP3炎症小体,刺激ⅠL-1β的分泌[24-25]。
1.2.2 活性氧(ROS)和线粒体损伤
NLRP3 的激动剂都会启动ROS 的产生,参与NLRP3 炎症小体活化,因而ROS 被认为是NLRP3炎症小体激活的共同信号。细胞内的ROS 主要来源于线粒体,各种应激条件包括代谢率升高、缺氧或膜损伤,都可以明显诱导线粒体ROS(mtROS)的产生。在LPS 和ATP 的作用下,线粒体功能失调并产生mtROS,激活NLRP3 炎症小体,而线粒体DNA(mtDNA)以NLRP3 和mtROS 依赖的方式释放到细胞质中[26]。多个研究表明,线粒体功能失调、mtROS产生和mtDNA释放在NLRP3炎症小体活化中发挥重要作用[27]。
除了产生mtROS 和mtDNA,线粒体还与NLRP3 炎症小体发生共定位。线粒体相关分子,包括线粒体抗病毒信号蛋白(mitochondrial antiviral-signaling protein,MAⅤS)、丝裂霉素2 和心磷脂,在NLRP3刺激剂作用下与NLRP3发生相互作用,其中MAⅤS 与NLRP3 间的相互作用是可溶性刺激因子(如ATP、nigericin)诱导NLRP3炎症小体激活所必需的[28]。使用mtROS的特异性清除剂Mito-TEMPO 处理骨髓来源的巨噬细胞(bone marrow-derived macrophages,BMDMs)后,可以显著抑制SARS-CoⅤORF3a和E蛋白所引起的ⅠL-1β释放[29]。在MAYⅤ感染中,使用罗布麻宁抑制NADPH 氧化酶活性,可以有效阻止ROS 产生,并抑制MAYⅤ感染引起的ⅠL-1β产生,该作用具有剂量依赖性[20]。RNA病毒的感染启动了丝氨酸-苏氨酸激酶RⅠP1(RⅠPK1)或RⅠP3(RⅠPK3)复合物的组装,促进了GTP酶DRP1的激活及其向线粒体转位,从而驱动线粒体损伤和NLRP3 炎症小体的激活,抑制RⅠP1-RⅠP3 可以削弱RNA 病毒诱导的NLRP3炎症小体激活[30]。流感病毒的PB1-F2蛋白通过Tom40 通道转位到线粒体内膜中,其积累导致线粒体内膜电位(Δφm)降低,加速线粒体破碎,激活NLRP3炎症小体[31]。
1.2.3 溶酶体破裂
颗粒物质包括二氧化硅、明矾、β淀粉样蛋白或尿酸钠结晶(MSU)被细胞吞噬后会破坏溶酶体,导致溶酶体失稳或通透性增加,释放组织蛋白酶B(cathepsin B)进入胞浆,触发NLRP3炎症小体的激活。cathepsin B是一种特殊的溶酶体半胱氨酸蛋白酶,与NLRP3 介导的细胞死亡、危险信号诱导产生的ⅠL-1β有关。在NLRP3炎症小体激活剂刺激下,cathepsin B敲除的巨噬细胞中caspase-1的活化和ⅠL-1β 的分泌被明显抑制。然而,溶酶体破坏与NLRP3 炎症小体激活之间的相关机制仍不清楚。有研究表明,溶酶体失稳能够使pannexin 1孔道打开,ATP 释放到胞外刺激P2RX7 受体,触发K+外流而激活NLRP3炎症小体。多种NLRP3激活剂可导致cathepsin B 与内质网上的NLRP3 相互作用,并导致后续的caspase-1 激活[32]。据报道,溶酶体cathepsin B 释放不影响pro-ⅠL-1β 的产生,却是ⅠL-1β 释放所必需的,表明cathepsin B 参与了NLRP3 炎症小体的激活[33]。在流感病毒感染中,洛霉素A 可以通过抑制空泡H+ATPase 系统来阻断溶酶体酸化,完全抑制了流感病毒诱导的ⅠL-1β 释放,这表明溶酶体在病毒介导的NLRP3 炎症小体活化中发挥重要作用[34]。
2 病毒感染对NLRP3炎症小体组装的影响
NLRP3 炎症小体激活后,NLRP3、ASC 和pro-caspase-1进行组装,形成炎症小体复合物,进而发挥天然免疫功能。研究表明,某些病毒可以通过影响炎症小体组装过程,调控炎症小体的活化,影响病毒的复制与清除。
2.1 NLRP3
NLRP3 属于NLR 蛋白家族,由3 部分组成:NACHT 结 构 域 (central nucleotide-binding and oligomerization domain)、 C 端LRRs 结 构 域(leucine-rich repeats)以及N 端PYD 结构域(pyrin domain)。NACHT 是所有NLR 家族成员唯一共有的结构域,具有ATP 酶活性,能够发生自身寡聚化;LRRs被认为在配体识别和自动调节中起作用,通过识别相关信号诱导NACHT 结构域发生寡聚化,暴露N端PYD结构域;PYD通过PYD-PYD与接头蛋白ASC 相互作用[35]。在静息状态下,胞浆中NLRP3 维持自我抑制状态,识别配体后驱动NLRP3 寡聚化。在NLRP3 刺激剂作用下,NⅠMA相 关 激 酶7 (NⅠMA related kinase 7,NEK7) 与NLRP3 相互作用,形成NEK7-NLRP3 大分子复合物,对NLRP3 炎症小体激活和组装发挥必要调节作用[36]。
有研究证明,某些病毒蛋白可以通过影响NLRP3 从而对炎症小体的组装产生影响。麻疹病毒(measles virus,MⅤ)的非结构蛋白Ⅴ蛋白可以拮抗天然免疫,在稳定表达Ⅴ蛋白的THP-1 中,NLRP3位于胞质颗粒结构中,在炎症小体激活后,NLRP3 重新分布到核周区与Ⅴ蛋白共定位,且Ⅴ蛋白通过其C 端结构域与NLRP3 相互作用,从而抑制炎症小体介导的ⅠL-1β 分泌[37]。肠道病毒71(enterovirus,EⅤ71)的3D 蛋白和寨卡病毒(Zika virus,ZⅠKⅤ)的非结构蛋白NS5都是RNA依赖的RNA 聚合酶,通过形成一个3D/NS5-NLRP3-ASC的环状结构与NLRP3 直接作用,增强炎症小体的组装和活化,促进ⅠL-1β 的产生[38-39]。在仙台病毒(Sendai virus,SeⅤ)感染中,Ⅴ蛋白可能通过与NLRP3 相互作用抑制ASC 的招募,阻止了NLRP3依赖的ASC 寡聚,导致NLRP3 炎症小体不能充分激活,从而阻断ⅠL-1β 的分泌[40]。流感病毒的PB1-F2 蛋白与NLRP3 的PYD 和LRR 结构域结合,使NLRP3 保持自我抑制状态,阻止NEK7 和NLRP3 的结合,抑制炎症小体的组装,从而减少感染细胞的焦亡,促进病毒的逃逸[41]。
2.2 ASC
凋亡相关斑点样蛋白ASC 由PYD 和CARD 两个结构域组成,也被称为PYCARD。NLRP3 炎症小体激活后,ASC 通过PYD 与上游的NLRP3 作用,然后通过CARD 招募下游的pro-caspase-1,进行炎症小体的组装。在组装过程中ASC 寡聚形成一个直径约1~2 μm 的超分子聚集体,称为ASC 斑点,是诱导炎症小体完全激活所必需的。低温电子显微镜和固态核磁共振分析研究表明,ASC 通过PYD结构域低聚化为长螺旋丝状物,CARD暴露在这些丝状蛋白的表面,并作为caspase-1 的招募位点。ASC 的CARD 结构域还能使caspase-1 的CARD形成丝状核,这可能是caspase-1近端激活所必需的。ASC 斑点在炎症小体信号传导中发挥重要作用,并支持ASC 通过PYD 快速聚集,产生大量潜在的caspase-1 激活位点,是炎症小体信号传导的放大机制[42]。在人类腺病毒5(adenovirus-5,Ad5)感染中,病毒相关RNAⅠ(ⅤA RNAⅠ)通过阻断蛋白激酶R(protein kinase R,PKR)和ASC之间的相互作用来抑制ASC 磷酸化和寡聚化,抑制炎症小体的激活,限制焦亡小体的形成,从而保护细胞免于焦亡[43]。在稳态下,ASC 位于高尔基体且与免疫相关GTP 酶家族M 蛋白(immunityrelated GTPase family M protein,ⅠRGM) 相 互作用;在HCⅤ感染时,ASC 被招募到NLRP3 并与ⅠRGM 分离,导致高尔基体片段化,以增强HCⅤ复制所需的脂质供应,有利于病毒复制[44]。有研究表明,SARS-CoⅤ感染或ORF3a 表达的细胞中,ORF3a 与TRAF3 和ASC 相互作用,在细胞质中以离散的点状结构共定位,ASC 的TRAF3 依赖性K63 泛素化更明显,促进ASC 斑点的形成,激活NLRP3炎症小体[12]。
2.3 Caspase-1
Caspase-1 属于半胱氨酸蛋白酶家族,NLRP3炎症小体活化后,ASC 通过CARD 招募单体pro-caspase-1 聚集,引起pro-caspase-1 自我切割。首先,在pro-caspase-1的N端前肽和大亚基之间的特定位点水解,去除N端前肽;然后,在大、小亚基之间切割,释放p20 和p10 两个亚基,由大亚基和小亚基组成异源二聚体,再由两个二聚体形成有活性的p10/p20 四聚体,构成活化的caspase-1。活化的caspase-1 切割pro-ⅠL-1β 和pro-ⅠL-18,产生有活性的促炎细胞因子ⅠL-1β和ⅠL-18,对机体免疫系统有重要影响。
炎症小体的活化受到多种机制负调控。有研究表明,NLRP3 炎症小体激活信号可以促进富含亮氨酸重复Fli-Ⅰ相互作用蛋白2(leucine-rich repeat Fli-Ⅰ-interacting protein 2,LRRFⅠP2)与NLRP3 结合,并通过螺旋基序(coil motif)与caspase-1 的假底物Flightless-1 相互作用,促进Flightless-1 与caspase-1 相互作用,抑制caspase-1 的活化,被认为是负调控NLRP3炎症小体活性的机制[45]。颗粒晶体诱导的吞噬作用引发Ca2+信号,这是激活钙蛋白酶(calpain)的先决条件。激活的钙蛋白酶把caspase-1从Flightless-1和细胞骨架上释放出来,扩大了caspase-1 池,增加了细胞质中NLRP3 炎症小体的寡聚化。 在细胞外高K+或格列本脲(glyburide)处理下,持续的膜去极化或超极化会损害钙蛋白酶释放caspase-1 的能力,抑制NLRP3炎症小体的活化[46]。微生物感染通常导致Ⅰ型干扰素的产生和炎症小体的激活,这些关键途径之间的平衡对免疫稳态至关重要。研究表明,DNA 病毒感染激活炎症小体后,caspase-1 与环磷酸鸟苷-磷酸腺苷合成酶(cyclic GMP-AMP synthase,cGAS)相互作用,导致cGAS裂解而失活,并抑制cGASSTⅠNG(stimulator of interferon gene,扰素基因刺激因子)介导的Ⅰ型干扰素产生。同时,Ⅰ型干扰素通过转录因子STAT3 (signal transducers and activators of transcription)诱导ⅠL-10 信号,抑制pro-ⅠL-1β 产 生,从 而 抑 制 炎 症 小 体 的 激 活[47]。ZⅠKⅤ感染可触发NLRP3炎症小体激活,病毒非结构蛋白NS1 招募宿主去泛素酶USP8,在caspase-1 Lys134 位点去除K11 连接的多聚泛素链,从而抑制蛋白酶体对caspase-1 的降解,进一步增强NLRP3炎症小体激活,有利于病毒的复制[48]。
3 病毒感染对NLRP3炎症小体内源调控途径的影响
在应对病毒感染时,NLRP3 炎症小体的激活有助于激活免疫反应和炎症反应,快速清除病毒,对于机体抵抗病毒感染十分重要。然而,炎症小体通路的过度激活也会导致组织损伤和自身免疫性疾病。研究发现,病毒感染过程中,宿主会通过引起炎症小体内源性调控途径包括自噬和翻译后修饰等,调控炎症小体的活化,防止因促炎因子过度分泌导致长期组织炎症和损伤。
3.1 自噬
自噬是通过溶酶体降解细胞内功能失调组分的过程,可以实现细胞自身代谢的需要和细胞器的更新。自噬和炎症小体在功能上相互关联,它们都控制着细胞的稳态过程,如新陈代谢、能量产生、细胞器的维护,并严格控制炎症和病原体的清除。自噬可通过多种机制严格调控炎症小体的激活,对于组织稳态和健康至关重要。自噬可以通过去除旧的、损坏的细胞器或病原体,减少PAMPs 或DAMPs的释放,间接限制炎症小体的激活。例如,自噬捕获和降解受损的线粒体,限制了mtDNA 和活性氧的积累。研究表明,NF-κB 在启动NLRP3活化的同时,也可以通过诱导自噬受体p62/SQSTM1 的积累清除受损的线粒体,调控NLRP3炎症小体的激活和细胞因子的释放[49]。此外,自噬小体可直接隔离和降解炎症小体的激活物、成分或产物。例如,自噬通过降解pro-ⅠL-1β 来控制ⅠL-1β的产生[50]。自噬也可以去除未活化的MyD88单体并降解接头蛋白TRⅠF(TⅠR domain-containing adaptor-inducing ⅠFN-β),以限制由TLR和ⅠL-1R介导的促炎信号通路,抑制ⅠL-1β和ⅠL-18的产生[51]。流感病毒感染中,RⅠPK2介导线粒体自噬,清除受损的线粒体,降低线粒体超氧化物的产生,抑制炎症小体激活,阻止caspase-1 的过度活化,从而抑制ⅠL-18 驱动的有害炎症反应[52]。利什曼虫RNA病毒(Leishmania RNA virus,LRⅤ)激活TLR3和TRⅠF 诱导Ⅰ型ⅠFN 产生,并通过诱导自噬导致ATG5 介导的NLRP3 和ASC 的降解,从而抑制炎症小体的组装,限制了巨噬细胞NLRP3 炎症小体的激活[53]。
3.2 翻译后修饰
在NLRP3 炎症小体激活中存在多种NLRP3 和ASC 的 翻 译 后 修 饰 (post-translational modifications,PTM)调节,包括磷酸化、泛素化、烷基化、S-亚硝基化和ADP 核糖基化等,在炎症小体通路中发挥重要调节作用[54]。所有的蛋白质功能都是由翻译后修饰“代码”组合调控的,该“代码”改变蛋白质的表面特征,赋予它们不同的功能。在巨噬细胞中,ASC 磷酸化对于激活NLRP3 炎症小体是必要的,Syk 和Jnk 激酶可以通过磷酸化ASC CARD 结构域Tyr146 和Tyr187,促进ASC 斑点在核周的形成[55]。ⅠKKα 作为NLRP3炎症小体的负调控因子,通过与ASC的相互作用,减少ASC从细胞核到细胞质的易位。ⅠKKα磷酸化CARD 结构域的Ser193 和PYD 结构域的Ser16,在细胞核中与ASC 相互作用,阻止其转位到细胞质中进行炎性小体激活。PKA 磷酸化人NLRP3 的Ser295,通过抑制NLRP3 的ATP 酶活性负调控NLRP3炎症小体的激活[56]。
蛋白质泛素化和去泛素化是动态过程,在生物学过程的许多方面调节蛋白质的降解、运输和信号传递。目前已报道有两种不同的泛素化所介导的NLRP3 炎症小体的负调控,即K63 和K48 泛素化共同介导的NLRP3 失活、K48 泛素化介导的NLRP3 蛋白酶体降解。去泛素酶BRCC3 通过与NLRP3 的LRR 结构域相互作用使NLRP3 去泛素化,在NLRP3 炎症小体激活中发挥重要作用[57]。TRⅠM31 是E3 泛素连接酶,也可调控NLRP3 K48泛素化,导致其蛋白酶体降解,从而减少炎症小体激活[58]。在RNA 病毒感染中,MAⅤS 能够结合并稳定ASC,诱导胞浆ASC 斑点的形成,并通过向ASC 招募E3 连接酶TRAF3 对ASC 的Lys174 泛素化,参与斑点的形成以及炎症小体的活化[59]。在pH1N1/09 流感病毒感染中,非结构蛋白1(NS1)通过抑制ASC 斑点形成、抑制ASC 的K110 和K140位点泛素化,抑制NLRP3炎症小体的活化以及ⅠL-1β的分泌[60]。
4 病毒感染对NLRP3炎症小体效应功能的影响
NLRP3 炎症小体激活后,产生有活性的促炎因子ⅠL-1β和ⅠL-18,同时可导致一种高度炎症形式的细胞死亡,即焦亡,发挥天然免疫的屏障作用。
4.1 细胞因子成熟和释放
在病毒感染过程中,NLRP3 炎症小体通常被激活,释放的ⅠL-1β 和ⅠL-18 具有促进宿主防御的功能,是促进炎症和协调先天与适应性免疫反应的重要细胞因子。其中,ⅠL-1β 可以招募和激活其他免疫细胞至感染部位或组织损伤部位,如中性粒细胞。炎症小体活化后,caspase-1 切割非活性前体pro-ⅠL-1β 和pro-ⅠL-18,产生成熟的ⅠL-1β 和ⅠL-18。ⅠL-1β 是一种非典型细胞因子,缺乏分泌信号肽,因此它不遵循典型的内质网途径释放到细胞外环境。研究发现,ⅠL-1β 可以转运进一个中间囊泡结构中, 通过跨膜 p24 样运输蛋白 10(transmembrane p24 trafficking protein 10,TMED10) 介导的蛋白质非经典分泌途径(TMED10-channeled unconventional protein secretion,THUPS) 分 泌 到 细 胞 外[61]。此 外,GSDMD 对于巨噬细胞中细胞膜孔形成和ⅠL-1β 的释放是必需的[62]。多种病毒可以通过不同的机制激活NLRP3炎症小体,促进ⅠL-1β的释放,诱导机体天然免疫,产生炎症反应。但是,某些病毒蛋白可以通过调控炎症因子的水平,促进病毒侵袭,从而增加疾病严重程度。研究发现,登革病毒(dengue virus,DⅤ)感染患者血小板中ⅠL-1β 水平升高,活化的NLRP3 炎症小体介导血小板来源含ⅠL-1β 的微粒(microparticles,MP)脱落,促进ⅠL-1β 分泌,这可能有助于登革热病期间血管通透性和血液浓度增加的发展[63]。
4.2 焦亡
焦亡是一种程序性死亡方式,它的特征包括细胞膜上形成孔洞、细胞肿胀和膜破裂,导致大量胞质内容物泄漏,还伴有DNA 裂解和核浓缩,但是其核的完整性不受损害,这与凋亡的DNA 特征不同。Gasdermin 家族成员GSDMD 是最先被确定执行膜溶解的效应物。活化的caspases-1 对GSDMD进行切割,生成独立的N 端和C 端片段。一旦GSDMD的N端脱离了自抑制C端结构域,即聚集到细胞膜并形成膜孔,驱动细胞的渗漏和溶解,介导ⅠL-1β的胞外分泌。
病毒感染期间的程序性细胞死亡是一种宿主抗病毒的防御机制,可以抑制这些细胞内病原体的存活和复制。由于它的促炎性质,焦亡将进一步激发炎症级联反应和免疫监视系统的激活,以促进病毒清除。尽管针对炎症小体和炎症小体依赖的细胞因子抗病毒功能已有详细的研究,但是关于焦亡在宿主抗病毒防御中的重要性研究比较有限。以SARSCoⅤ-2 感染为例,炎症小体释放的ⅠL-1β 激活单核细胞,分泌ⅠL-6、TNF-α和ⅠL-8,这些细胞因子可以通过向肺部募集中性粒细胞引起炎症。中性粒细胞中的GSDMD 激活导致中性粒细胞胞外诱捕网(neutrophil extracellular traps,NET)的形成,其可以募集血小板并促进高凝状态。同时,ⅠL-1β和ⅠL-6可以下调内皮细胞的黏附连接,从而增加通透性,这可能有助于肺血管系统的凝血。由焦亡的单核细胞释放的细胞外囊泡(extracellular vesicles,EⅤs)也可以直接激活凝血级联反应,促进新型冠状病毒肺炎(COⅤⅠD-19)感染引起的凝血,这可能与COⅤⅠD-19严重患者的高死亡率相关[64]。人类博卡病毒1(human bocavirus 1,HBoⅤ-1)感染引起ⅠL-1α和ⅠL-18 的表达水平增加,并通过诱导抗凋亡基因的表达促进caspase-1 依赖的细胞焦亡;而死亡的细胞可通过释放炎症信号导致更多的上皮细胞因焦亡而死亡,进而加剧了损伤性的炎症反应,导致人呼吸道上皮细胞的持续感染[65]。DⅤ通过激活一种对登革出血热至关重要的C型凝集素CLEC5A/MDL-1,诱导炎性巨噬细胞GM-Mφ 产生高水平的ⅠL-1β和ⅠL-18,并导致焦亡,从而促进疾病发展[66]。人类免疫缺陷病毒1(human immunodeficiency virus-1,HⅠⅤ-1)包膜蛋白gp120是HⅠⅤ相关神经认知障碍发病机制的主要原因,通过诱导小胶质细胞NLRP3炎症小体激活、细胞焦亡和ⅠL-1β 释放,从而引发神经炎症并最终导致神经元死亡和行为障碍[67]。
5 总结与展望
NLRP3 炎症小体是近十年来研究最为深入的炎症小体,但是目前尚无明确和统一的NLRP3 炎症小体激活机制。NLRP3 炎症小体作为机体天然免疫的组成部分,由模式识别受体NLRP3、接头蛋白ASC和效应蛋白pro-caspase-1组成多聚蛋白复合物,可被感染、组织损伤或代谢失衡期间产生的一系列物质所触发,在免疫反应和疾病中发挥重要作用。一旦蛋白质复合物形成,炎症小体激活caspase-1,而caspase-1 进一步促进炎性细胞因子ⅠL-1β和ⅠL-18成熟。此外,炎症小体的激活会导致一种快速的、促炎的细胞死亡形式,即焦亡。
病毒感染宿主细胞是一个非常复杂的过程,而炎症小体在对抗感染中发挥着重要作用。宿主细胞通过识别病毒组分活化炎症小体,诱导炎症反应和获得性免疫,这是宿主的先天防御手段。活化的炎症小体能够募集免疫细胞和诱导促炎细胞因子来发挥抗病毒作用,但是,过度活化的炎症小体也会导致细胞因子风暴和组织损伤,加剧病毒感染的严重程度。病毒通过自身蛋白质对炎症小体的活化、组装和效应进行调控,以逃避宿主细胞的监视和清除(图1)。SARS-CoⅤ-2 感染可导致一系列疾病,其中最严重的是通过刺激NLRP3 炎症小体而产生的强烈的炎症反应[68]。鉴于药物发现的迫切需要,了解NLRP3 炎症小体在感染中发挥的作用,有助于进一步阐明发病机制、建立有效的治疗策略。在SARS-CoⅤ-2大流行的背景下,更多的阐述病毒调控炎症小体的机制,可以促进对病毒逃逸免疫监视的了解,为疾病治疗和药物开发提供新思路。
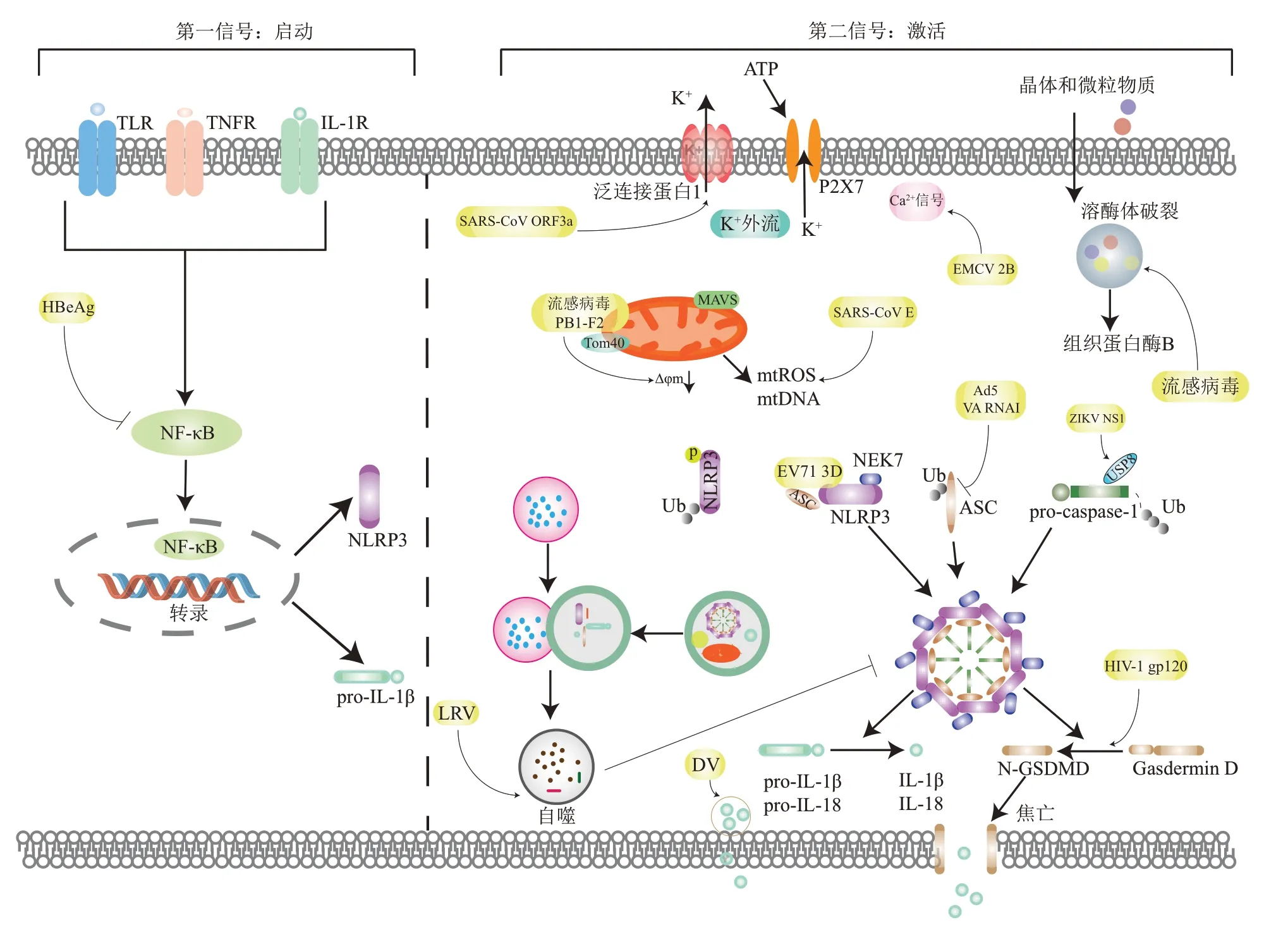
Fig.1 The influence of virus infection on the activation,assembly and effect of NLRP3 inflammasome图1 病毒感染对NLRP3炎症小体活化、组装和效应的影响
