Cr,Ce共掺杂调控V3O4半金属材料自旋极化机制的第一性原理研究
2022-07-17牛永,王盼
牛 永, 王 盼
(陕西科技大学 文理学院, 陕西 西安 710021)
0 引言
半金属材料是指在费米能级处,其中一个自旋方向的能带具有非零的带隙呈现出非金属性,而在另一个自旋方向的能带贯穿费米面呈现出金属性,因此兼具金属性和半导体的特征[1-3].因为半金属材料在费米面附近只有一种自旋取向的电子,从而具有极高的自旋极化率(理论上100%),在自旋半导体器件中具有非常广阔的应用前景[4-6].


图1 理想立方相2×2×1 V3O4
国内外关于V3O4材料的磁性研究较少,因为通常氧化钒化合物的化学式可以表示为VnO2n-1(其中n为整数≥1)和VnO2n+1(其中n为整数≥2),然而V3O4无法用整数的化学式来表示[7].在实验中通过溶剂热法合成出尺寸为4.8 nm的立方相V3O4量子点,在室温表现出没有磁滞的超顺磁性且阻挡温度为32 K[8].从应用的角度考虑,一个好的自旋电子学材料应该具有高的居里温度同时要具有大的磁化强度,因此如何将V3O4材料的居里温度提高,是此半金属化材料应用于自旋半导体器件所面临的一个重要问题,而理论研究可为提高V3O4材料的磁学性质提供有效的理论指导和方法.
居里温度决于自旋交换耦合作用,而自旋交换耦合常数决定于体系自旋反平行(EAFM)与平行状态(EFM)下系统的总能量差值ΔE=EAFM-EFM[9-17].正的ΔE表示系统为铁磁基态,且ΔE越大说明铁磁状态越稳定,对应的居里温度越高,室温铁磁性材料的能量差值ΔE应大于室温热能(25 meV)[18].而掺杂可改变半导体中载流子的种类并引入杂质能级,进而改变自旋交换耦合作用,为调控材料居里温度提供了有效手段[19,20].Cr元素与主体材料中的V元素都同属于过渡金属,其电子态为3d54s1,具有不同的3d电子数.稀土族Ce元素具有特殊的4f电子结构,掺杂后将引入强的4f电子局域自旋,按照RKKY磁性机制Ce离子巡游的6s电子自旋极化后作为媒介实现4f电子局域自旋之间的交换相互作用[21,22].这两种元素的共掺杂可在V3O4材料中实现不同类型的磁性作用,将带来一种更为全新复杂的铁磁性和居里温度形成机制.因此,理论研究过渡金属Cr与稀土Ce元素不同位置的共掺杂,对于调控V3O4材料的居里温度具有重要意义.
因此,针对目前立方相V3O4半金属材料铁磁性居里温度低的问题,本论文基于第一性原理计算采用过渡金属Cr与稀土Ce不同位置的共掺杂来调控V3O4材料的自旋极化和自旋交换耦合作用,揭示不同Cr和Ce共掺杂对V3O4材料的铁磁性和居里温度的影响机制,为提高V3O4半金属材料的磁学性质提供理论指导和方法,同时也为自旋电子学的研究提供理论基础.
1 计算方法和参数
采用第一性原理Quantum ESPRESSO软件[23]来计算V3O4的自旋态密度,此计算是基于密度泛函理论(DFT)[24]以及投影缀加平面波方法(PAW)贋势[25,26]得到.由于V3O4材料中过渡金属V原子中3 d电子具有强关联相互作用,需要在原来的DFT基础上添加平均场Hubbard项,这里采用了包含了电子关联效应的了GGA+U方法.根据之前V3O4的文献[8]报道,V元素施加U值为4.5 eV.由于Cr原子的3d电子和Ce原子的4f电子都具有强关联相互作用,当引入Cr和Ce掺杂时,需要在原来的DFT基础上添加平均场Hubbard项,这里采用了包含了电子关联效应的了GGA+U方法.对Ce掺杂施加2-6 eV的U值,经计算可知,不同U值对应Ce单掺杂V3O4材料的自旋上下的态密度图都发生了劈裂.当Ce元素的U值为5 eV时,Ce单掺杂V3O4体系的能量最低,因此对Ce元素施加5 eV的U值.对Cr单掺杂V3O4材料分别施加1-4 eV的U值,不同U值对应Cr单掺杂V3O4材料的自旋上下的态密度图基本一致,且都发生了劈裂.当Cr元素的U值为1 eV时,Cr单掺杂V3O4体系的能量最低,因此对Cr元素施加1 eV的U值.
为研究不同Cr、Ce共掺杂对V3O4材料自旋态密度的影响,对立方相V3O4原胞进行2×2×1扩胞,并引入(a)Cr和Ce掺杂都占据A位置四配位的V原子,(b)Cr和Ce掺杂都占据B位置六配位的V原子,(c)Cr和Ce掺杂分别占据A位置四配位和B位置六配位的V原子,(d)Cr和Ce掺杂分别占据B位置六配位和A位置四配位的V原子,形成不同Cr和Ce共掺2×2×1立方相V3O4体系(V22CrCeO32).对于结构优化和静态计算,布里渊区采用Monkhorst-Park方法产生3×3×6的K点网格.计算的截断能是570 eV,能量和力收敛标准分别是10-5eV和0.01 eV/Å.
2 V3O4的自旋极化机制研究
2.1 理想V3O4的自旋态密度和自旋电荷密度
由理想V3O4自旋态密度图2(a)可知,自旋向上的价带被费米能级穿过显示出金属性,且此电子态主要来源于V原子的3d轨道与O原子2p轨道的耦合作用;而自旋向下的态密度在费米能级附近处没有电子态显示出半导体特性,因此V3O4总体呈现出半金属特性,在费米能级处只有一种自旋方向的电子态,为100%高自旋极化.进一步由其自旋电荷密度分布图2(b)可知,其自旋电荷密度主要分布于V原子上,计算得到的磁矩为54.926μB.为了研究理想V3O4的铁磁稳定性,本文计算了反铁磁(AFM)和铁磁(FM)状态下的能量差值ΔE(ΔE=EAFM-EFM).由表1可知,ΔE值为正,说明反铁磁状态的能量大于铁磁状态,磁基态类型为铁磁性.
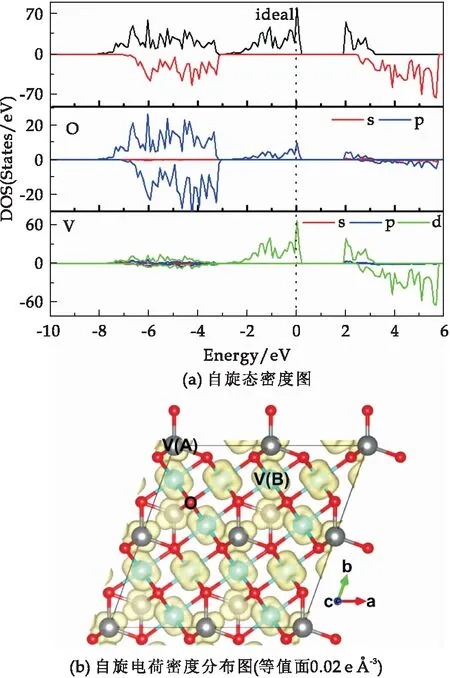
图2 理想2×2×1立方相V3O4超胞(V24O32)

表1 理想2×2×1 V3O4超胞体系反铁磁和铁磁 状态的能量(E)、总磁矩(M)、能量差值(ΔE)和 磁基态类型
2.2 Cr、Ce共掺杂对V3O4的自旋态密度和自旋电荷密度的影响研究
图3(a)和(b)分别时Cr、Ce共掺杂都占据四配位A位的V原子时的2×2×1 立方相V3O4体系的自旋态密度图和自旋电荷密度图.当Cr和Ce掺杂都占据四配位A位时,由图3(a)的自旋态密度图可知,与理想的V3O4体系相比,其自旋向上的态密度基本不变,但是在自旋向下态密度图中的价带和导带的带隙中引入了两个杂质能级,降低了费米能级附近的自旋极化程度,且此杂质能级主要来源于O原子的2p轨道和Ce原子的4f轨道的耦合作用.进一步由图3(b)的自旋电荷密度图可知,与理想的V3O4体系相比,Ce原子上的自旋电荷密度远小于原位置处V原子的自旋电荷密度,其他原子上的自旋电荷密度分布基本不变,因此整体的自旋电荷密度分布降低,与减小的总磁矩51.494μB相一致.
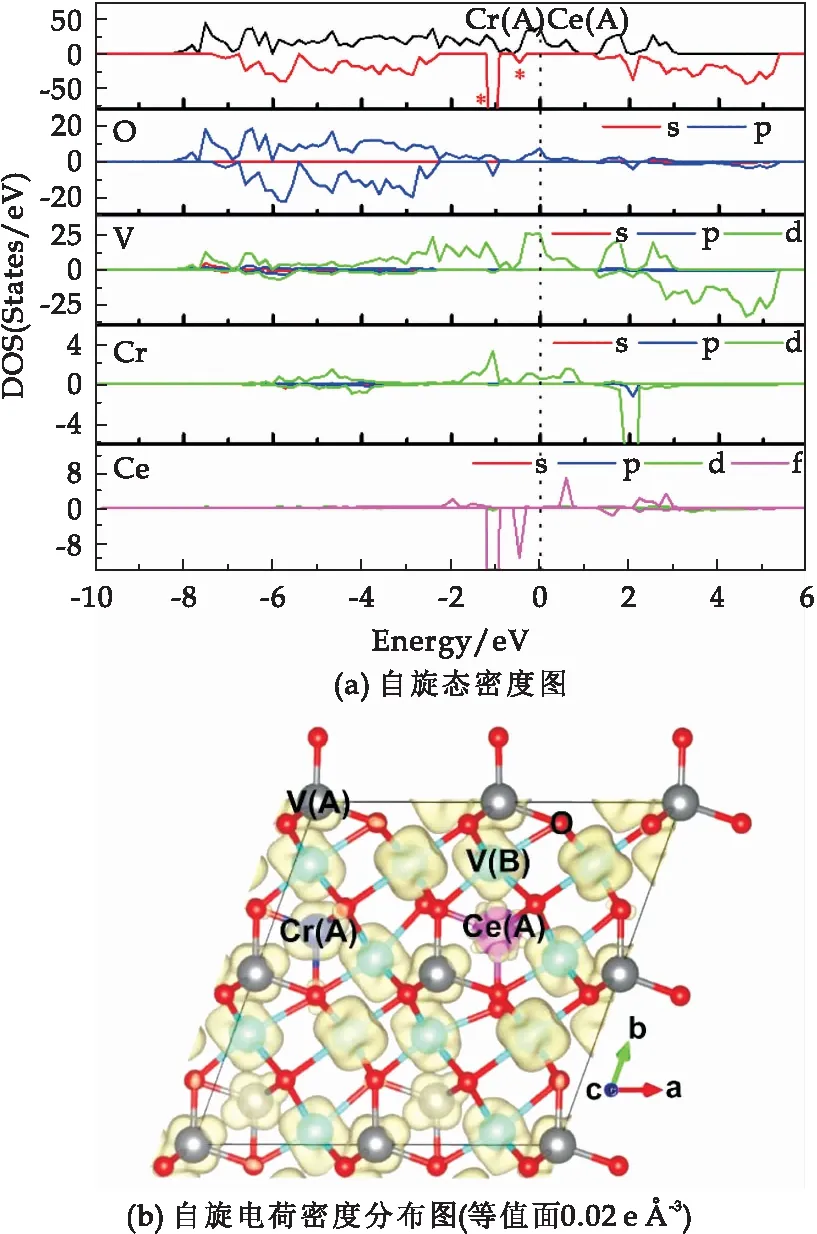
图3 Cr和Ce掺杂都占据A位四配位的 2×2×1立方相V3O4体系
当Cr和Ce掺杂都占据六配位B位时,由图4(a)的自旋态密度图可知,与理想的V3O4体系相比,其自旋向上的态密度基本不变,但是在自旋向下态密度图中的价带顶引入了杂质能级,降低了费米能级附近的自旋极化程度,并且此杂质能级主要来源于O原子的2p轨道和Cr原子的3d轨道的耦合作用.进一步由图4(b)的自旋电荷密度图可知,Ce原子的自旋电荷密度分布远小于原位置处V原子的自旋电荷密度分布,且Ce原子周围的V原子的自旋电荷密度分布也降低了,因此整体的自旋电荷密度分布降低,与减小的总磁矩47.280μB相一致.


图4 Cr和Ce掺杂都占据B位时六配位的 2×2×1 V3O4体系
当Cr和Ce共掺杂分别占据A位四配位和B位六配位时,由图5(a)的自旋态密度图可知,与理想的V3O4体系相比,其自旋向上的态密度基本不变,但是在自旋向下态密度图中价带和导带的带隙中引入了三个杂质能级,降低了费米能级附近的自旋极化程度,并且这些杂质能级主要来源于O原子的2p轨道和V原子的3d轨道的耦合作用.进一步由图5(b)的自旋电荷密度图可知,Ce原子上的自旋电荷密度远小于原位置处V原子的自旋电荷密度分布,且Cr掺杂原子周围的V原子的自旋电荷密度分布也降低了,因此整体的自旋电荷密度分布降低,与其减小的总磁矩45.335μB相一致.
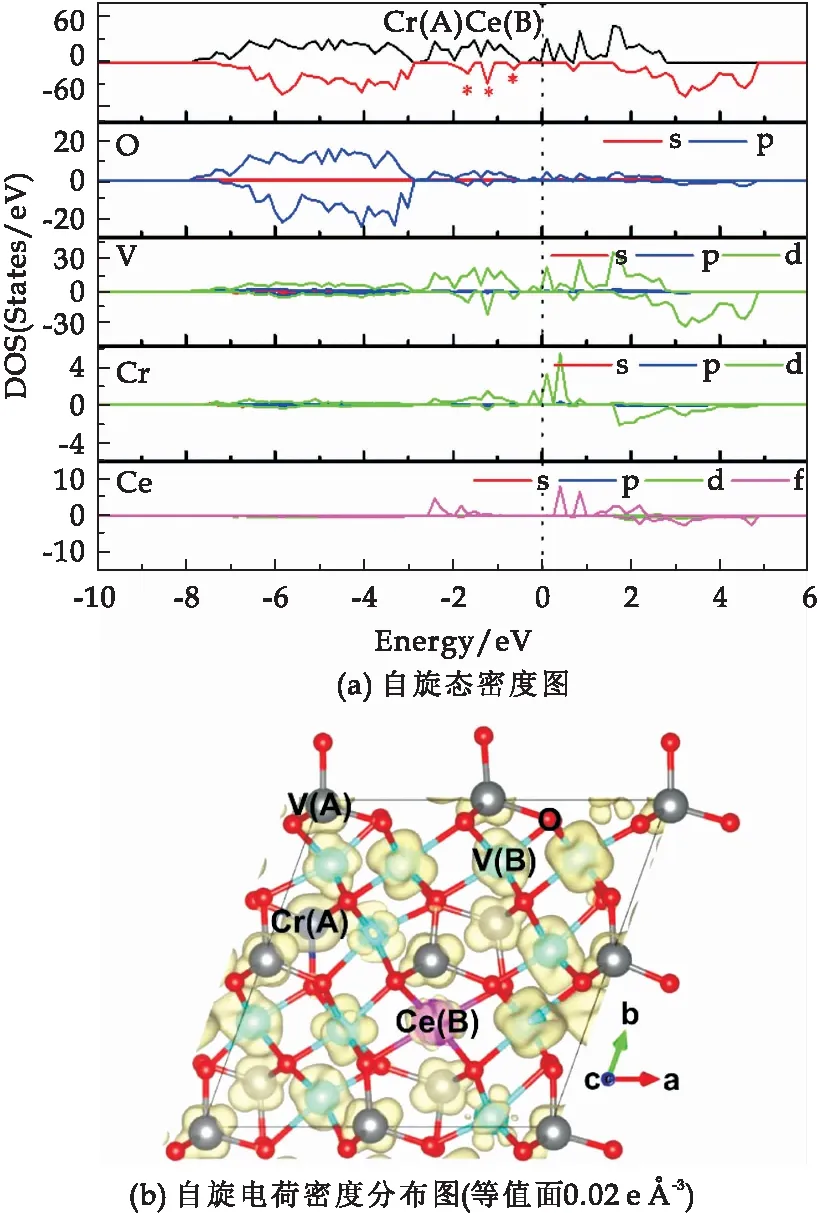
图5 Cr和Ce共掺杂分别占据A位四配位和 B位六配位的2×2×1立方相V3O4体系
当Cr和Ce共掺杂分别占据B位六配位和A位四配位时,由图6(a)的自旋态密度图可知,与理想的V3O4体系相比,其自旋向上的态密度基本不变,但是在自旋向下态密度图中价带和导带的带隙中引入了两个杂质能级,降低了费米能级附近的自旋极化程度,并且这些杂质能级主要来源于O原子的2p轨道和Cr原子的3d轨道的耦合作用.进一步由图6(b)的自旋电荷密度图可知,Ce原子的自旋电荷密度分布很小,远远低于原位置处V原子的自旋电荷密度分布,因此整体的自旋电荷密度分布降低,与其减小的总磁矩47.617μB相一致.

图6 Cr和Ce共掺杂分别占据B位六配位和 A位四配位的2×2×1 立方相V3O4体系
为了研究不同位置的Cr和Ce共掺杂对V3O4体系的铁磁稳定性的影响,我们计算了反铁磁(AFM)和铁磁(FM)状态下的能量差值ΔE(ΔE=EAFM-EFM).表2分别列举了Cr和Ce掺杂都占据A位(Cr(A)Ce(A)),Cr和Ce掺杂都占据B位(Cr(B)Ce(B)),Cr和Ce掺杂分别占据A位和B位(Cr(A)Ce(B)),以及Cr和Ce掺杂分别占据B位和A位(Cr(B)Ce(A))的2×2×1立方相V3O4超胞的反铁磁和铁磁状态的能量差值(ΔE),以及磁基态类型.
由表2可知,对于这四种不同位置Cr和Ce共掺杂的2×2×1立方相V3O4超胞的ΔE值都为正,说明反铁磁状态的能量大于铁磁状态,则磁基态类型均为铁磁性.且与理想的V3O4相比,当引入Cr和Ce共掺杂分别占据A和B位时,其ΔE值降低,说明铁磁态稳定性和居里温度减小;当引入其他三种位置的Cr和Ce共掺杂时,其ΔE值都明显增大,说明铁磁态稳定性和居里温度极大提高;特别是当Cr和Ce共掺杂都占据A位,其ΔE值为原来的33倍,能最大提高其铁磁稳定性和居里温度.
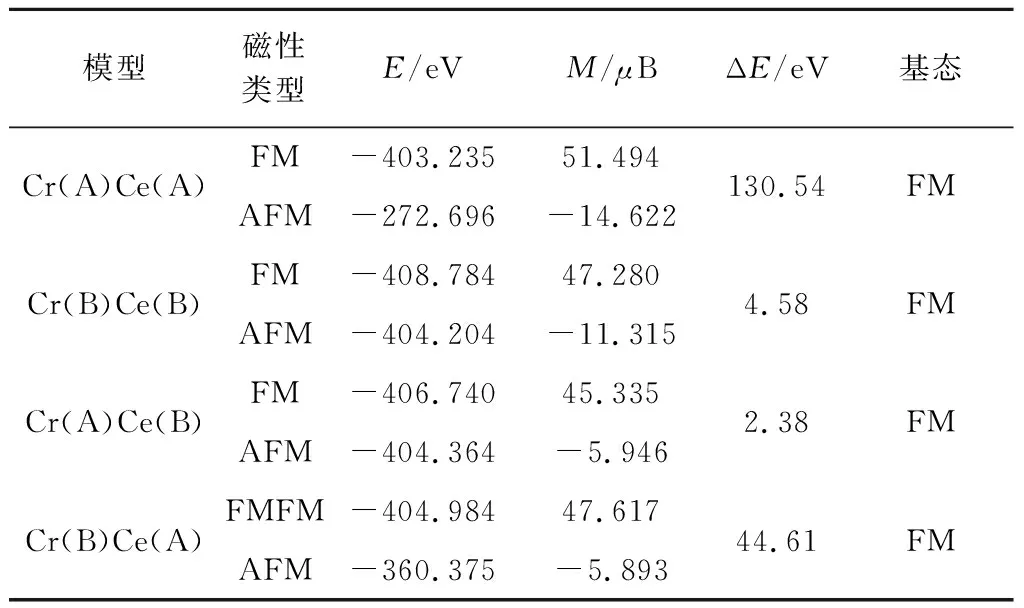
表2 理想2×2×1 V3O4超胞体系,以及Cr、Ce 共掺杂立方相2×2×1 V3O4超胞体系反铁磁 (AFM)、铁磁(FM)状态的能量(E)、总磁矩(M)、 能量差值(ΔE)和磁基态类型
3 结论
本论文采用第一性原理计算研究了不同位置的Cr和Ce共掺杂对V3O4自旋极化的影响机制.计算结果表明理想的V3O4为半金属性,且其磁基态为铁磁性.不同位置的Cr和Ce共掺杂都降低了V3O4的自旋极化率和总磁矩.当引入Cr和Ce共掺杂都占据A位V原子,Cr和Ce都占据B位V原子,以及Cr和Ce分别占据B和A位V原子这三种共掺杂时,其ΔE值都明显增大,说明铁磁态稳定性和居里温度极大提高.特别是当Cr和Ce共掺杂都占据A位,其ΔE值为原来的33倍,能最大化提高其铁磁稳定性和居里温度.以上研究为提高V3O4的铁磁稳定性和居里温度提供了有效途径,进一步促进了V3O4在自旋电子学中的应用.
