Clouston综合征家系的病例报告1例
2022-06-15曲春燕王锋马登科赵敏王晓力孙毅梁凤和
曲春燕王锋马登科赵敏王晓力孙毅梁凤和
1国家儿童医学中心首都医科大学附属北京儿童医院保健中心(北京100045)
2山东省康复研究中心(济南 250109)
3北京博奥晶典生物技术有限公司(北京 102206)
4中国听力语言康复研究中心(北京 100029)
5首都医科大学附属北京同仁医院耳鼻喉科头颈外科(北京 100730)
1 病例资料
患者男,7岁,以自幼听力障碍伴头发及指(趾)甲发育异常就诊并进行基因检测,临床诊断Clouston综合征。出生未做听力筛查,2岁半时因不会说话发现听力障碍,当地诊断为双侧极重度感音神经性耳聋并验配助听器。5岁时进行右耳人工耳蜗植入手术开始语言康复。图1是术前5岁时裸耳听力和术后7岁时助听听阈(右耳人工耳蜗,左耳助听器)。耳蜗术前听力学和影像学结果:双耳ABR 100dB nHL未引出;双耳畸变耳声发射各频率均未引出;双耳声导抗226Hz鼓室图均为As型;颞骨CT和MRI未见异常。患者的一般情况、发育和智力无明显异常。头发稀疏、软、颜色偏浅呈棕色,没有明显片状缺失(图2A)。指(趾)甲发育不良,外端1/3-1/2部分粗糙增厚,颜色棕黄不透明(图2B、2C和2D),汗腺和皮脂腺分泌正常,掌跖部未见明显异常。其姐姐、父亲有类似的毛发和指(趾)甲特点(图2E),母亲无类似表现。询问父系家族史,爷爷有毛发和指(趾)甲类似表现,奶奶、姑姑和叔叔均无异常,患者家系图见图3A。用遗传性耳聋基因芯片对先证者的4个常见耳聋基因9个位点进行芯片筛查,包括GJB2(c.35delG、c.176del16、c.235delC、c.299_300delAT),SLC26A4(IVS7-2A>G、c.2168A>G),线粒体基因12S rRNA(mt.1555A>G、mt.1494C>T),GJB3(c.538C>T),结果为阴性,后续对先证者进行全外显子测序,未发现GJB2和GJB3基因的变异。全外显子测序发现GJB6基因杂合突变c.31G>A(p.G11R)/-,将该突变与OMIM数据库收录的明确致病相关基因进行比对,怀疑为致病的分子病因。通过Sanger测序对先证者、姐姐和父母样本进行了验证,父亲和姐姐均携带该突变,测序峰图结果见图3B,母亲为野生型,遗传方式符合常染色体显性遗传。患者是该家系中唯一伴有听力障碍的病例。患者人工耳蜗术后进行了1年半的康复训练,听觉能力进步明显,由开始的47%逐步提高到90%,语言能力进步相对缓慢,在康复1年半之后才由2岁提高到3岁(表1)。5年后电话随访,家长描述患者和姐姐随着年龄的增长,头发密度增加,但发色依然偏浅,发质比较脆容易断。患者现在普通小学就读,可以与家长和同学进行日常沟通交流。
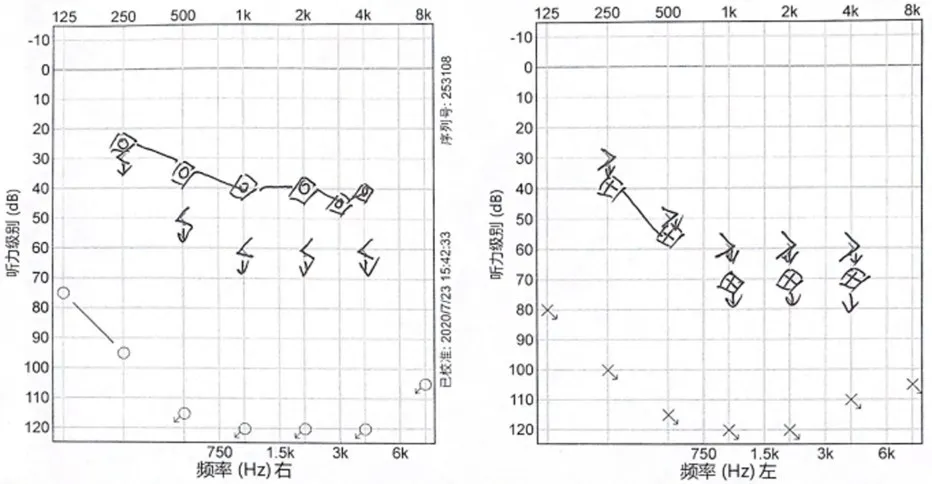
图1 术前5岁裸耳听力和术后7岁助听听阈:右耳人工耳蜗,左耳助听器Fig.1 Unaided hearing threshold at 5-year-old before cochlear implant surgery and aided hearing threshold at 7-year-old:right ear with CI,left ear with HA.

图2 患者及其父亲的头发和指(趾)甲表现.A:患者与旁边正常儿童头发相比,明显的稀少和发色浅;B;患者双手指甲;C:患者双手大拇指近照;D:患者双脚趾甲;E:患者父亲双手指甲。Fig.2 Features of hair and nail in patient and his father.A:patient has typical thin hair and light color compared with the normal child next to him;B:Fingernails of patient;C:Close look at patient’s thumbnails;D:Toenails of patient;E:Fingernails of patient’s father.

图3 患者的家系图和测序峰图.A:家系图,箭头所指为毛发和指(趾)甲发育异常并伴有耳聋的先征者;B:患者即先征者GJB6基因Sanger测序峰图,箭头所指为c.31G>A(p.G11R)/-杂合突变Fig.3 Family tree and Sanger sequencing.A:Proband with nail dystrophy,thin hair and congenital deafness(arrow),his older sister,his father and grandfather have nail dystrophy,thin hair without congenital deafness;B:Sanger sequencing results of proband with heterozygous mutation c.31G>A(p.G11R)/-in GJB6 gene(arrow).

表1 患者人工耳蜗术后不同时间段听觉能力和语言能力的康复评估结果Table 1 Assessment of auditory and language skills in different time points of rehabilitation after cochlear implant
2 讨论
Clouston综合征是Clouston于1929年首先在加拿大法国裔中发现并报道了该病,并因此命名[1]。也称为有汗性外胚层发育不良(hidrotic ectodermal dysplasia,HED),临床主要表现为指(趾)甲营养不良、毛发稀疏、掌跖角化过度,部分患者伴有耳聋、白内障、斜视、智力障碍和骨骼改变等,汗腺和皮脂腺功能正常。该病致病基因是编码缝隙连接蛋白30(Cx30)的GJB6基因,为常染色体显性遗传,目前发现该基因的5种突变均为错义突变,包括p.G11R[2,3]、p.V37E[4]、p.D50N[5]、p.A88V[6]和 p.N14S[7],导致肽链N端一个氨基酸被替换。
缝隙连接通道是细胞间的一种特殊细胞膜间通道,进行相邻细胞内分子量小于1 kDa的离子、亲水性的代谢物质的直接转运。目前为止,缝隙连接蛋白家族有21个成员[8],分布广泛,除红细胞、精母细胞、横纹肌细胞外[9,10],几乎在所有的组织细胞都有表达。Cx26、Cx29、Cx30、Cx31和Cx43在人耳蜗都有表达,但分布有所不同:Cx26和Cx30在耳蜗Cortis器的支持细胞、耳蜗侧壁血管纹的基底细胞和中间细胞、螺旋韧带和螺旋缘的纤维细胞间有共表达,是构成耳蜗缝隙连接的主要蛋白成员;Cx29表达在包绕耳蜗螺旋神经节细胞的Schwann细胞上;Cx31表达在耳蜗侧壁螺旋韧带的III型纤维细胞上;Cx43表达在耳蜗骨性结构上和骨内侧壁细胞上[11]。GJB2基因(编码Cx26)和GJB6基因(编码Cx30)突变导致了近一半的先天性非综合征性耳聋病例,是遗传性耳聋的主要致病基因[12-16],也与综合征性耳聋密切相关。GJB2和GJB6基因突变导致耳聋的病理机制有不少假说,其中主要一个是缝隙连接的功能障碍引起内耳K+离子循环障碍,导致K+离子在毛细胞周围的细胞间隙潴留而引起毛细胞的中毒损伤和凋亡[17,18]。也有学者发现敲除Cx26的小鼠主要是耳蜗发育异常导致的先天性耳聋,并与缝隙连接介导的miRNA细胞间基因调控有关,因此对内耳K+离子循环障碍的假说提出质疑[19]。至少7种缝隙连接蛋白,包括Cx26,Cx30 and Cx43在人类皮肤有表达,对于上皮细胞分化和伤口愈合起重要作用。编码Cx26、Cx30、Cx30.3和Cx31的某些基因突变常常导致听力障碍合并各类皮肤疾病,呈现常染色体显性遗传方式[15]。
GJB6基因突变可能导致Cx30蛋白结构异常、转运异常、分布异常等,但GJB6基因如何引起不同疾病的发病机制至今不清。目前为止,只有p.G11R、p.V37E、p.D50N、p.A88V和p.N14S这5个突变与Clouston综合征有关。p.G11R和p.A88V突变在不同人群中都有发现[16],p.G11R突变是最常见的致病突变[20],也是本文家系的致病突变。通过体外细胞学和分子生物学的研究探讨其致病机理,有研究表明p.A88V或p.G11R突变的表达会引起Ha-CaT细胞的凋亡[21]。大鼠角质细胞表达p.V37E或p.A88V突变导致Cx30蛋白异常折叠而聚集在细胞浆的内质网,导致细胞膜上的表达减少。只引起非综合征性耳聋的p.T5M突变,细胞膜上的表达与野生型相似。p.V37E和p.A88V突变引起细胞凋亡,即使同时表达野生型Cx26或Cx30也不能挽救细胞的死亡,提示这两种突变有显性负性效应(dominant-negative effect)[22]。另外一个研究发现p.V37E或p.A88V突变的Cx30在人体受损和正常的皮肤中,表达没有差异。体外研究p.V37E或p.A88V突变在HeLa细胞膜上与野生型Cx30共表达并形成细胞间通道,形成的通道电生理特性与野生型Cx30没有明显区别,但形成的功能获得性(gain of function)半通道导致细胞内ATP向细胞外介质泄漏,增加的ATP会作为旁分泌信使,影响了角化细胞的增殖和分化[23]。
Clouston综合征非常罕见,发病率约十万分之一。本文家系中的三位突变携带者均有指(趾)甲营养不良和毛发稀疏的表现,但都没有掌跖角化过度,其中先证者伴有听力障碍,另外两位听力正常。印度一个同为c.31G>A(p.G11R)致病突变的5代大家系的10例携带者,均表现指(趾)甲营养不良,2例无毛发稀疏,均无听力障碍[20]。2013年报道的一个中国家系,8名p.G11R突变携带者都有相似的临床表现,包括指(趾)甲营养不良、毛发稀疏和掌跖角化过度,但没有听力障碍[24]。2013年报道的一个三代台湾家系,p.G11R突变受累者均有指(趾)甲畸形,无毛发缺失和掌跖角化过度[25]。2014年报道一例p.G11R突变散发病例,13岁中国男童,毛发和指(趾)甲受累,掌跖部未见明显异常,无听力障碍[26]。p.A88V突变也是相对比较多见的GJB6基因致病突变,河南一252人的大家系中,对携带p.A88V突变的37人的临床资料进行分析,100%都有毛发受累、92%的人有指(趾)甲营养不良,57%的人有掌跖角化过度[27],p.A88V突变也在其他中国Clouston家系中被发现[28,29],均无听力障碍。有研究发现p.A88V突变可以减少耳蜗缝隙连接通道的表达,对耳聋有长期的保护作用,所以该突变导致的Clouston综合征不伴有听力障碍[30]。但这种保护性作用只是体外细胞水平的研究结果,并不能反映人体内的病理生理过程,不伴有耳聋可能只是表型差异的原因。携带p.A88V突变的转基因小鼠的短纯音和短声ABR波潜伏期和幅度与野生型小鼠无区别,但阈值有变化,纯合突变小鼠的低频ABR阈值升高、高频ABR阈值和DPOAE值比杂合或者野生型小鼠要低[31]。也有文献报道p.A88V突变导致的Clouston综合征伴轻度耳聋[32],该病例为一位日本24岁女性,先天性完全无毛发,包括头发、睫毛、眉毛和身体其它部位,伴指(趾)甲营养差、掌跖角化过度、轻度畏光和耳聋。耳聋为先天性双侧轻度感音神经性耳聋,但言语正常。该病例除了携带GJB6基因p.A88V杂合突变还携带GJB2基因p.V27I杂合变异,作者分析尽管p.V27I杂合变异是多态性,畏光和耳聋的表型可能与之有关。这个分析结论还需要更多基础研究和临床资料的支持。角膜炎-鱼鳞病-耳聋(KID)综合征多为GJB2基因突变导致,一例KID综合征伴毛发缺失患者携带GJB6基因的p.V37E突变,提示KID和Clouston综合征在分子病因和表型上有重叠和交叉[33,34]。
Clouston综合征的临床表型多变,一些患者仅有头发或指(趾)甲的异常,有可能被漏诊。本研究首次在GJB6基因p.G11R突变导致的Clouston综合征患者中发现耳聋情况,为该突变的遗传咨询和本文患儿及其姐姐的婚育提供了产前诊断的依据。
