一例非综合征型耳聋家系中POLR1D新突变位点的发现及分析
2022-06-15尹晶晶刘章张肖林徐琪钧王延飞赵洪春李长叶张如意刘秀珍
尹晶晶刘章 张肖林徐琪钧王延飞赵洪春李长叶张如意刘秀珍
1滨州医学院附属医院耳鼻咽喉头颈外科(滨州 256600)
2滨州医学院附属医院麻醉科(滨州 256600)
3滨州医学院附属医院医学研究中心(滨州 256600)
耳聋是当今世界常见的感觉障碍之一,它极大地损害患者的身心健康,全世界每1000名新生儿中就有1例耳聋患者[1]。近50%的耳聋是由遗传因素引起,在发达国家,约有6%~8%的人有遗传性耳聋[2,3]。遗传性耳聋分为综合征型和非综合征型,综合征型耳聋涉及身体其他表型,如Usher、Waardenburg和Treacher Collins综合征等;非综合征型耳聋可进一步分为常染色体隐性(75%~80%)、常染色体显性(15%~25%)、X染色体连锁和线粒体耳聋(1%~2%)[4,5],现已鉴定的单基因非综合征型耳聋基因有124个。
POLR1D位于13号染色体,编码的POLR1D蛋白是一种进化保守的蛋白,最初在酵母中被鉴定为核糖核酸聚合酶I(PolI)的亚基,后来被证明也是核糖核酸聚合酶 III(PolIII)的一个亚基[6]。PolI和PolIII都参与核糖体RNA和小RNA的合成,POLR1D突变会导致核糖体发生障碍[7]。核糖体生物发生的中断通常导致胚胎发育或成人体内平衡的障碍,人类POLR1D突变导致Treacher Collins(TCS)综合征(TMIM:613717)[8]。TCS综合征是一种罕见的先天性颌骨颜面发育不全综合征,听力学主要表现为外耳或中耳畸形导致的传导性耳聋,每50,000例活产中就有1例发生[9]。
本研究是通过对一例非综合征型遗传性耳聋家系进行分子遗传学分析,发现一个新的耳聋基因突变位点,此突变可能与该家系非综合征型耳聋相关。本研究为阐明耳聋新的发生机制,拓宽对遗传性耳聋疾病的认识提供参考。
1 材料与方法
1.1 研究对象
本研究对象来自滨州医学院附属医院耳鼻咽喉科门诊的一例非综合征型耳聋家系,已获得家系成员知情同意,研究方案经滨州医学院附属医院伦理委员会批准实施(伦理批准号:LW-020)。收集家系成员临床资料,采集外周血,并对家系成员进行听力(音叉试验)及体格检查,对耳聋患者做纯音听阈测定检查。
1.2 基因组DNA提取及基因检测
采用血液基因组DNA提取试剂盒(TIANGEN,DP318)提取外周血的基因组DNA。采用Sanger测序对四个耳聋基因(GJB2、SLC26A4、GJB3和MTRNR1)的21个常见突变位点进行检测;如未见突变,则利用高通量测序对这四个常见耳聋基因进行全测序;如四基因全序列无致病突变,则进行全外显子检测(WES)(北京诺禾致源科技股份有限公司)和耳聋基因Panel-V3(北京迈基诺医学检验所)检测,并对检测结果进行生物信息学分析及验证,在 UCSC(http://genome.ucsc.edu/)上分析突变位点的保守性。
1.3 小鼠耳蜗RNA提取及Real-time PCR
取C57BL/6J小鼠E12.5(精栓出现第12.5天)、E16.5、D0(出生当天)、D7、D14、D21、D28、2M(2月龄)、12M((12月龄,雄性)耳蜗组织。Trizol试剂(Invitrogen,美国)提取组织RNA,逆转录试剂盒(PK0498,TaKaRa,日本)将RNA反转录为cDNA。Real-time-PCR采用SYBR Green II q PCR SuperMix(TaKaRa,日本)试剂,BioRad CFX Real-time-PCR仪。本研究使用引物均采用Primer5软件设计,内参为Gapdh,序列见表1。
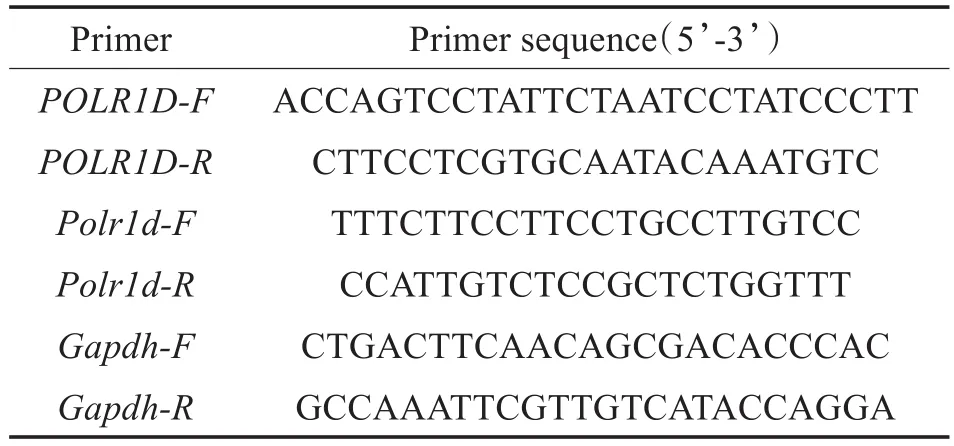
表1 引物序列Table 1 Primer sequence
2 结果
2.1 家系成员临床症状及听力结果
家系图见图1,先证者(Ⅳ-12),男,31岁,为佩戴助听器来我院就诊,自述20岁时出现双耳听力下降,无耳鸣、眩晕及其他不适,纯音听阈测定结果显示感音神经性聋,双耳听损基本对称见图2A。其母亲(Ⅲ-7)48岁时出现左耳听力下降,54岁时纯音听阈测定结果显示左耳听力显著下降,特别是高频区4000-8000HZ听力完全丧失,右耳较好,只在高频区轻度下降,见图2B;先证者外祖母及姨祖母(Ⅱ-1、Ⅱ-5)也有听力障碍,已去世,耳聋发生年龄不详;先证者2位姨母及表姐(Ⅲ-3、Ⅲ-9、Ⅳ-4),分别在9岁、20岁、17岁时出现双耳听力下降,纯音听阈测定结果为双耳感音神经性耳聋,双侧听损基本对称,见图2C、D、E;Ⅴ-10偶有耳痛,已治愈,听力正常,其余成员听力正常。耳聋患者均无耳毒性药物应用史、噪声接触史及头部外伤史,面部发育正常,无其它器官发育不良。

图1 非综合征型耳聋家系图谱。箭头:先证者Fig.1 Family map of non-syndromic deafness.The arrow:the propositus
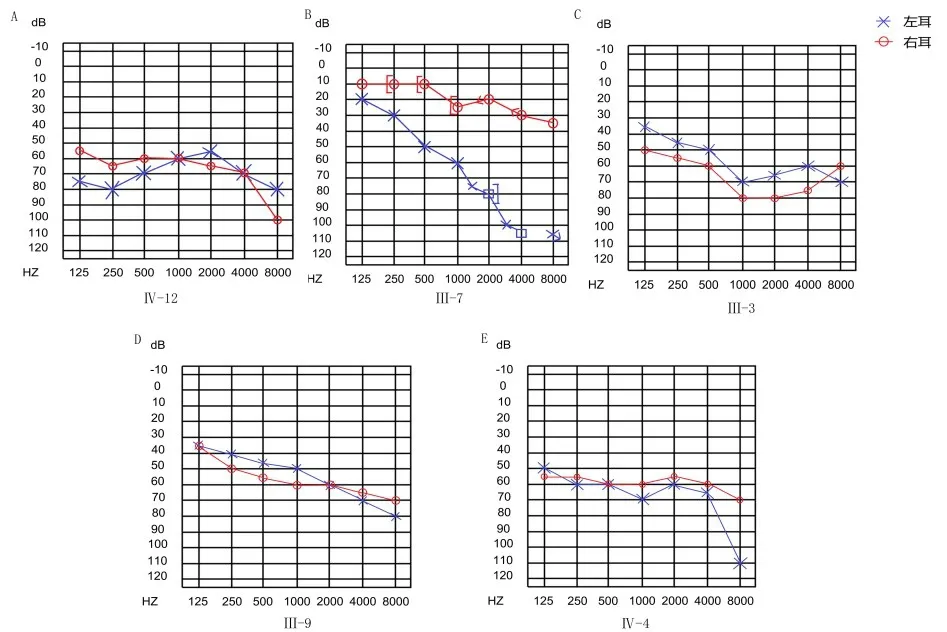
图2 非综合征型耳聋家系患者纯音听阈测定结果Fig.2 Test results of pure tone hearing threshold in families with non-syndromic deafness
2.2 常见耳聋基因和WES检测未发现致病突变
我们对先证者Ⅳ-12样本进行四个常见耳聋基因的21常见突变位点检测和四个耳聋基因全测序结果均未发现基因突变(结果未出示)。然后对1例正常(III-5)和3例患者(III-9、IV-4、IV-12)的样本进行WES检测,对筛选出的候选基因与感音神经性耳聋表型进行相关性分析并排序,对排名前20位的基因结合其有害性进一步评价,选出相关系数和有害性较高的11个基因,具体信息见表2。然后对这些突变进行一代测序验证,结果未发现与耳聋表型共分离的突变(结果未出示)。
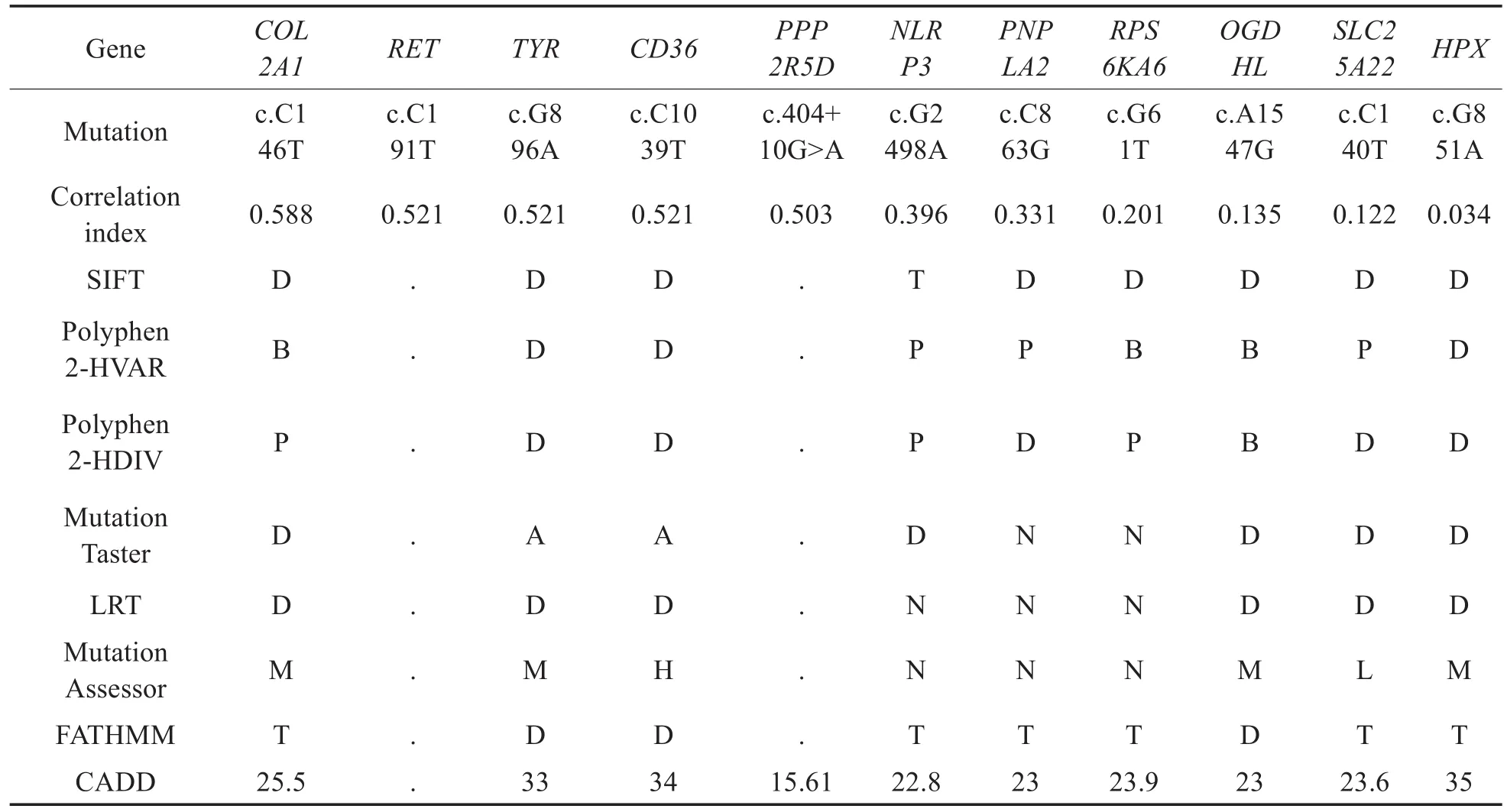
表2 候选基因相关性及有害性分析Table 2 Correlation and harmfulness analysis of candidate genes
2.3 耳聋基因Panel-V3检测发现一个新的POLR1D突变
耳聋基因Panel-V3是利用高通量技术对迄今发现的170个耳聋基因外显子及附近内含子进行检测,针对性强,不容易漏掉与耳聋密切相关的突变。由于WES检测未发现与该家系患者共分离的基因突变,因此我们对先证者(IV-12)进行耳聋基因Panel-V3检测,重点检测耳聋相关基因。结果共发现7个耳聋基因的8个突变,对检测结果进行测序验证,发现POLR1D2号外显子c.52A>G(p.M18V)突变与本研究家系的耳聋症状共分离见表3,基因突变测序结果,见图3。

图3 POLR1D突变位点的测序结果。Fig.3 Sequencing results of POLR1D mutation site.
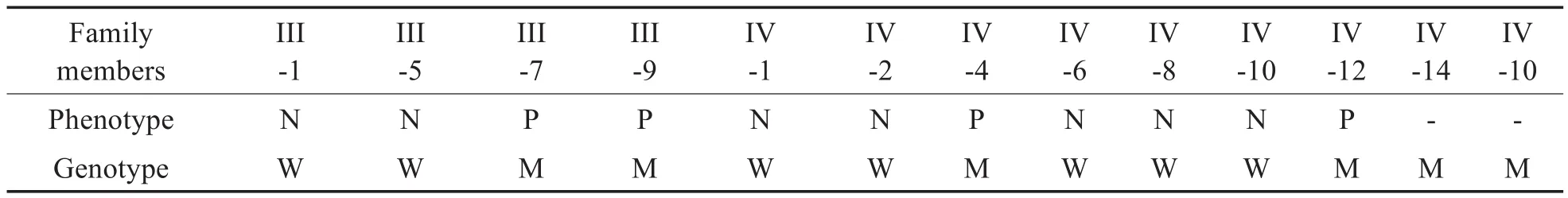
表3 一代测序验证结果和家系成员表型Table 3 The first generation sequencing verified the results and the phenotypes of the family members
2.4 POLR1D同源蛋白第18位甲硫氨酸(M18)在多个物种的高度保守
我们对不同物种POLR1D的同源蛋白氨基酸序列进行了比较,结果发现见图4红色矩形框内所示,POLR1Dc.52A>G(p.M18V)突变位点所编码的氨基酸在灵长总目(包含灵长类和啮齿类)、劳亚兽总目等多个物种中有高度保守性(见图4)。

图4 不同物种POLR1D同源蛋白氨基酸序列比对Fig.4 Amino acid sequence alignment of POLR1D homologous proteins of different species
2.5 Polr1dmRNA在不同发育时期小鼠耳蜗组织中的表达
我们用Real-time PCR方法检测了不同发育时期的C57BL/6J小鼠耳蜗Polr1dmRNA的表达,结果显示Polr1dmRNA在各个发育时期的小鼠耳蜗中均有表达,特别在12月龄小鼠耳蜗中的表达比胚胎期和年轻小鼠明显升高,见图5。

图5 Polr1dmRNA在小鼠不同时期耳蜗组织中的表达,横坐标表示小鼠发育时期或年龄,纵坐标表示mRNA的相对表达量,计算公式为 2-(CTPolr1d-CTGapdh),n=3。Fig.5 Expression of Polr1d mRNA in cochlea tissues of mice at different stages.The abscissa represents the different developmental stages of the mouse.The ordinate represents the relative expression of mRNA.2-(CTPolr1d-CTGapdh),n=3.
3 讨论
本研究对一例非综合征型耳聋家系进行了听力学检查和遗传学检测,发现POLR1D一个新突变(c.52A>G),此突变位点在多个物种间高度保守,且Polr1d的mRNA在各发育时期小鼠耳蜗中均有表达,推测此突变与该家系非综合征型耳聋有一定关系。
POLR1D编码PolI和PolIII中一个亚单位,PolI与PolIII参与的核糖核酸转录是核糖体生物发生的起始[10]。研究表明,POLR1D突变的斑马鱼胚胎在受精后9至10天内死亡[11]。POLR1D敲除对早期小鼠胚胎发生有重要影响,POLR1D缺失会导致严重DNA损伤、细胞增殖延迟和胚胎发育受损,每种情况均能导致胚胎死亡。人POLR1D突变可引起TCS综合征,其典型特点是双侧对称性下睑裂、颧骨发育不全、小颌和外耳畸形,约50%的人有传导性耳聋,通常由听骨畸形和中耳腔发育不全引起[12]。POLR1D突变引起非综合征型感音神经性耳聋迄今未见报道。TCS可由POLR1D、POLR1C和TCOF1突变引起[13],TCS患者中93%以上是由TCOF1突变引起,有报道一例TCS相关的TCOF1新突变家系,患者表现为TCS综合征,但具有相同突变的其父亲仅表现听力障碍[14],说明TCS相关基因突变也可引起单纯的耳聋症状。Shin-ya Nishio et al检测了Polr1d在耳蜗的表达,发现Polr1d在小鼠耳蜗科蒂氏区(基底膜)、侧壁区(血管纹、螺旋韧带和螺旋凸)、螺旋神经节区和螺旋缘(主要是齿间细胞区)等耳蜗多个部位表达,其中在螺旋神经节中比耳蜗其它部位的表达水平高3.2倍[15],螺旋神经节的功能是将声音信号传递给中枢神经系统,其受损会引起感音神经性耳聋。本研究中,我们检测了从胚胎期到12月龄小鼠耳蜗Polr1dmRNA的表达,发现在不同时期小鼠耳蜗中均有表达。较比年轻小鼠,在12月龄小鼠耳蜗中表达明显增高,提示Polr1d在耳蜗中可能有一定功能,且随着年龄增长,对该基因需求增大。这与本研究耳聋家系中患者表现有一致之处,患者在幼年听力正常,随着年龄增加,对该基因的需求也越大,如果该基因因突变而导致其蛋白产物某些性质的衰减,将会影响其功能发挥,最终出现听力减退。因此POLR1D突变除在TCS中引起中耳发育不全而导致传导性聋外,该基因突变还可能会影响耳蜗功能进而导致非传导性聋。本研究首先采用WES,验证了相关性和有害性排名较前的11个突变位点,但未发现与疾病共分离的突变。进而采用耳聋基因Panel进行针对性的检测,发现7个基因的8个突变,验证发现只有POLR1Dc.52A>G(p.M18V)与耳聋症状共分离,且该突变所在位点在多个物种中有很高的保守性。说明该位点对于维持POLR1D蛋白的功能非常重要,其突变可能会产生某种有害性。TCS综合征累及面部及外耳中耳发育,是一种严重的疾病,而本研究中发现的突变只是引起耳聋这一症状,表现较TCS轻,且并非传导性耳聋而是感音神经性聋,发病时间较晚。生信分析表明POLR1Dc.52A>G(p.M18V)其有害性和保守性并非所有突变中最强的一类突变,因此该突变并没有导致表型严重的TCS综合征,而只是成年后的单纯的感音神经性聋。综上所述,我们推测POLR1D突变位点c.52A>G(p.M18V)可能是本研究中非综合征型耳聋家系的致病因素,这是POLR1D突变首次与非综合征型感音神经性耳聋相联系。当然,要想确定二者因果关系,还需进一步验证,比如收取更多的耳聋样本,检测是否有POLR1D突变;制备动物模型,观察POLR1D相同位点的突变是否引起单纯的感音神经性聋。
此外,本家系中患者听力在幼年时均正常,一般在成年后发病,而家系第Ⅴ代成员年龄较小,未到发病年龄,因此无法统计其基因型和表型的关系。而Ⅳ-14(31岁)及其儿子(Ⅴ-10,9岁)均携带POLR1D突变,但都无耳聋症状,推测Ⅳ-14可能遗传了其母亲的背景基因,发病年龄较晚,因此,Ⅳ-14和Ⅴ-10二者的听力还需长期随访。在家族性遗传病中,由于成员间基因背景不同,即使携带同一致病基因,其表型往往也有差异。本家系三代成员均出现耳聋患者,且耳聋表型较为一致,提示为家族性聋,但Ⅲ-7双耳听损不对称,且发病较晚,与家系中其他患者差别较大,其原因可能由于遗传异质性所致。当然具体机制还有待进一步研究。
WES对全基因组外显子检测,筛选范围大,在发现疾病新的致病基因方面有强大优势,但也会因信息量太大而难以筛到关键突变[16]。耳聋基因Panel则是对耳聋基因进行检测,针对性强,但检测范围仅限于已知耳聋基因。本研究中我们首先进行WES检测,对候选突变进行验证后并未得到疾病共分离突变,当然我们可再选择其它突变继续分析,但工作量太大,难以进行一一验证。因此我们又采用耳聋基因Panel-V3进行检测,缩小筛选范围,期望找到致病突变。结果发现POLR1Dc.52A>G(p.M18V)与耳聋症状呈共分离。该突变在WES中也检测到,但生信分析表明POLR1Dc.52A>G(p.M18V)其有害性和保守性并非所有突变中最强的一类突变,因此没有列为候选突变,这是WES漏选的重要原因。耳聋基因Panel-V3与WES结果进行对比,发现除MYO1E突变外其余均与WES结果一致,说明两种检测结果均是可靠的,只是由于检测范围不同而各有优势,同时也说明即使WES检测后,Panel-V3检测也是有意义的。
总之,本研究在一例非综合征型耳聋家系中发现了POLR1D一个新突变位点c.52A>G,该突变可能是非综合征型耳聋的致病因素,这与以往报道的POLR1D突变引起的TCS不同,该发现为研究新的耳聋机制提供依据,扩展对聋病的认识。
