高电压钴酸锂电池的研究进展
2022-03-30程梅笑万广聪申海鹏郭营军李新丽
程梅笑,万广聪,申海鹏,郭营军,2,李新丽,2
(1.湖州昆仑亿恩科电池材料有限公司,浙江湖州 313103;2.香河昆仑化学制品有限公司,河北廊坊 065400)
钴酸锂(LCO)在所有锂离子电池正极材料中具有体积比能量高,工作电压范围宽,压实密度高,理论比容量大,且LCO 特殊的α-NaFe2层状结构可以实现Li+的快速迁移及稳定循环;但是,LCO 材料的实际比容量(140 mAh/g,Li1-xCoO2,x≈0.5,~4.2 Vvs.Li/Li+)只有理论值(274 mAh/g,Li1-xCoO2,x≈0.5,~4.2 Vvs.Li/Li+)的60%[1]。研究表明,通过提高电池的充电截止电压,可以大大提高LCO 正极材料的比容量以及能量密度,然而随着Li+的不断脱嵌,导致LCO 从六方晶相(O3相)到单斜晶相的不可逆相变[2-3]。此外,在高电压下LCO 材料界面与电解质间的副反应通常会导致LCO 电池容量下降及循环性能不稳定,从而限制了高电压LCO 电池的商业应用。为了充分发挥LCO 材料的应用价值,研究者进行了大量的研发工作,主要包括LCO 正极材料的改性及电解质添加剂的筛选。本文将着重介绍高电压LCO 电池正极材料的改性方法、电解质的优化方案及其中涉及的机理分析。
1 正极材料的研究进展
在锂离子电池充电到高截止电压的过程中,LCO 晶体结构经历了多种相变(H1 到H2,~3.9 V,绝缘体-金属转变;M1,~4.1 V;H3,~4.2 V,有序-无序转变;M2,~4.55 V;O1),导致晶体向c和a轴各向异性膨胀和收缩[4-5]。反复经历上述过程后,LCO 材料不可逆相变(例如,H2 到M1,M1 到H3,H3 到M2)增多,导致锂离子电池的容量衰减严重[6]。
1.1 掺杂法
体相元素掺杂可以改变LCO 材料原子级晶格结构,例如调节晶格缺陷比例,重排阳离子,重新分布电荷和电子结构,是提高LCO 材料结构稳定性最广泛使用的方法[7]。掺杂的元素分为过渡金属(Cu,Ni,Mn,Ti,La 等)及非过渡金属(Mg,Ca,Al,Si 等)两大类[8-9]。在晶体结构中掺杂元素通过占据锂离子或者钴离子的空间位置,达到稳定脱嵌锂晶体、扩大晶格间距及支撑层状结构的效果,进而抑制正极材料钴离子溶出及提升循环寿命[10]。
现阶段,研究者通过多种元素的协同掺杂进一步提升LCO 的电化学性能。Zhang 等[11]通过Ti-Mg-Al 共掺杂实现了LCO 电池(vs.Li/Li+)在4.6 V 下的稳定循环。在锂离子电池中,以0.5C倍率100 次循环后,仍实现了174 mAh/g 的高可逆放电比容量(与第二个循环相比,容量保持率86%)。研究表明,Mg 和Al 原子已成功掺杂到LCO 晶格中,从而改变了(去)锂化过程中的相变行为。Mg 的掺杂还可以增加材料的电子电导率。相反,即使微量的Ti 也不能完全掺入LCO 晶格中。Ti 在晶界和表面的偏析,一方面改变了样品颗粒的微观结构,这有利于整个Li+的扩散和均匀的内部应变分布;另一方面,抑制了氧的活性并使其稳定在高充电电压材料的表面。图1为三元素掺杂的空间及数量分布。电极材料的完美设计不仅需要探究掺杂元素的种类,也需要探究掺杂元素的用量。Xu等[12]探究了多元素(Al,Ti 和Mg)掺杂LCO 材料中Mg 用量对LCO 电化学性能的影响,结果表明,过量的Mg 掺杂不利于LCO 材料内部离子的动力学。这是由于过量的Mg 在LCO 材料表面形成了约2 nm 的MgO 涂层以及在内部随机形成MgO颗粒,导致Li+在晶体中脱嵌困难且锂离子电池放电平台降低。在综合考虑改性材料的循环性能(25 ℃,0.1C,200 次,容量保持率96%)、比容量(190.2 mAh/g)和动态性能(0.2C、0.5C、1C、2C以及0.2C,每种倍率5 个循环,最终容量保持率95%)的基础上,LCO 材料中掺杂0.12%(质量分数)的Mg 最适宜。Song 等[13]认为高电压LCO 材料中的多元素掺杂通常会由于晶界处的元素偏析而引起多晶化,这些元素偏析可以实现相邻初级粒子之间的稳固连接和缓冲,抑制了晶界处裂解和形成裂纹,并提供长期的稳定结构。
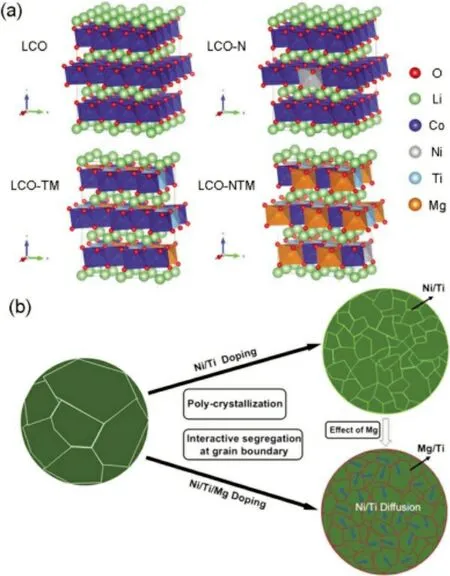
图1 (a)掺杂元素之间相互作用的理论计算与(b)多种元素掺杂的影响[13]
掺杂后的LCO 材料也有不稳定因素存在:(1)掺杂元素与电解质接触面积较大,导致严重的副反应;(2)掺杂元素各向异性膨胀,导致次级颗粒破裂;(3)掺杂元素在电解质中溶出。
1.2 包覆法
表面包覆法可以有效稳定LCO 材料界面,抑制不良反应进而提高高电压下锂离子电池的电化学性能[14]。表面包覆的材料通常具有高机械强度及化学稳定性,主要包括金属氧化物(例如:Al2O3,CuO,Y2O3),金属氟化物(例如:MgF2,CeF3,AlF3)和金属磷酸盐(例如:AlPO4,Li3PO4)等[15-16]。图2 为LCO材料包覆方法及示例。这些包覆材料通过在LCO 表面形成两相复合物(固溶体)而产生稳定的晶界,使去锂化状态下LCO 材料热稳定性增强并隔绝与电解质之间的副反应;但是LCO 的可逆容量降低,循环时电荷转移阻抗增加[17]。
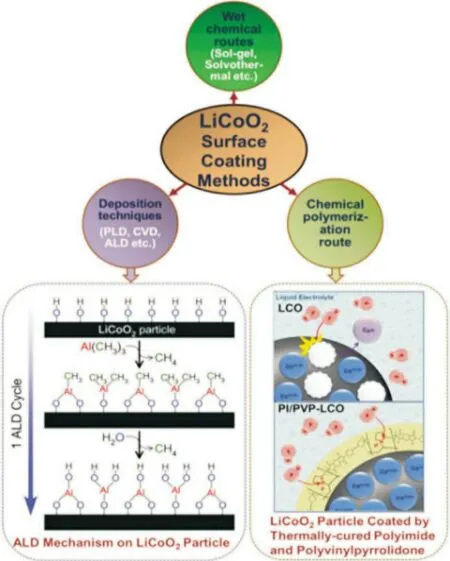
图2 LiCoO2的表面包覆方法流程图以及示例[17]
因此,研究者们为解决上述问题进行了实验探究。Cheng 等[18]提供了一种具有多层结构的表面改性方法,即在LCO 外层包覆富锌表面涂层,岩盐相缓冲层和表面梯度Al 掺杂层,实现了LCO 电池在4.6 V 下的稳定循环,并且修饰后的涂层抑制了LCO 材料与电解质的界面反应及阻抗的增长。材料表面准外延层生长现象表明,多层结构显著降低了LCO材料与表面涂层之间的晶格失配,增强了富锌外层的稳定性。此外,无序的岩盐相层和Al 表面掺杂也增强了结构稳定性。所有这些协同作用促使LCO 在4.6 V 下稳定循环,在500次循环后容量保持率为65.7%。一些研究人员认为,涂层可以抑制LCO 材料结构从H2 到M1 的相变,但是其他研究人员认为,包覆涂层不能抑制相变,但可以使LCO 材料晶相更具可逆性。Hu 等[19]利用湿化学法综合元素掺杂和表面包覆的优点,对LCO 材料进行了Ba 和Ti 二元混合改性,处理后得到LiCoO2(LCO@BT),即LCO 表面上形成的稳定的固态Li-Ti-Co-O 薄膜,同时掺杂少量BaTiO3和TiO2颗粒。研究表明,改性层可促进表面Li+扩散,降低电荷转移阻抗,并保护LCO 免受表面副反应引起的腐蚀。根据原位NMR 表征,在第一次电化学脱/嵌锂之后,发现LxCO@BT(0.98<x<1)中晶体O3-I型可逆转变,并且在4.5 V 的高截止电位下,改性材料初始放电比容量为190.5 mAh/g;0.2C,100 次循环后,容量保持率高达90.29%,放电比容量为180.4 mAh/g。
但是在实际工业生产过程中,受原料成本、制造环境及加工工艺等方面的影响,所面临的问题则是均匀包覆LCO 材料表面。
2 电解质的研究进展
高电压下,LCO 层状结构由于过度脱锂而变得不稳定,从而诱导Co4+溶解在液体电解质中;电解质在Co4+催化及高电势双重作用下被氧化分解生成大量气体。此外,溶解的Co4+迁移到负极并沉积在表面,导致电解质进一步还原分解并消耗活性Li+。常规的电解质碳酸酯类溶剂在Co4+的催化作用下往往达不到理论氧化电势(5 V,vs.Li/Li+)而在低电势(≤4.5 V,vs.Li/Li+)下就被氧化分解[20]。因此,开发具有较高的氧化电位,同时又可以抑制LCO 表面Co4+溶出的电解质,是高电压LCO 电池对电解质材料重要的要求。
2.1 电解质溶剂
理想状态下,锂离子电池负极的电化学势需要低于电解质溶剂的最低未占据分子轨道(LUMO)能级,正极的电化学势需要高于电解质溶剂的最高未占据分子轨道(HOMO)能级。因此高电压锂离子电池的进一步发展,需要电化学窗口大于5 V 的电解质溶剂。非对称无环砜基电解质溶剂已成为具有高电化学稳定性(>5.0 V,vs.Li/Li+)的最新电解质溶剂之一。Wu 等[21]通过前线分子轨道理论计算发现乙基甲基砜(EMS)、四亚甲基砜(TMS)和乙基乙烯基砜(EVS)具有相对较好的氧化稳定性。这三种砜从高到低的氧化稳定性为EMS、TMS 和EVS,氧化分解电位都在5 V 以上。图3 为锂盐和砜溶剂的电子云结构。除了砜类溶剂外,氟代溶剂也是一种良好的高电压电解质溶剂,例如氟代线性碳酸酯,氟代环状碳酸酯,氟代醚等[22]。氟原子具有很强的电负性和弱极性,氟取代氢会有效提高溶剂的氧化分解电压,氟化后的溶剂通常比原始溶剂的抗氧化能力高,浸润性好,符合高电压电解质溶剂的要求[23]。不过氟化后的溶剂对锂盐的溶解度降低,在氟代溶剂中往往需要添加助溶剂。Yan 等[24]将乙酸乙酯氟化后制备出DFEAc,由于氟原子具有很强的吸电子能力,因此提高了EA 的氧化耐受能力,并且DFEAc 还继承了EA 的低粘度,同时起到了高电压溶剂及助溶剂的作用;具有新型电解质的锂离子电池(vs.Li/ Li+)在4.6 V 下经过100 次循环后实现容量保持率89.23%。
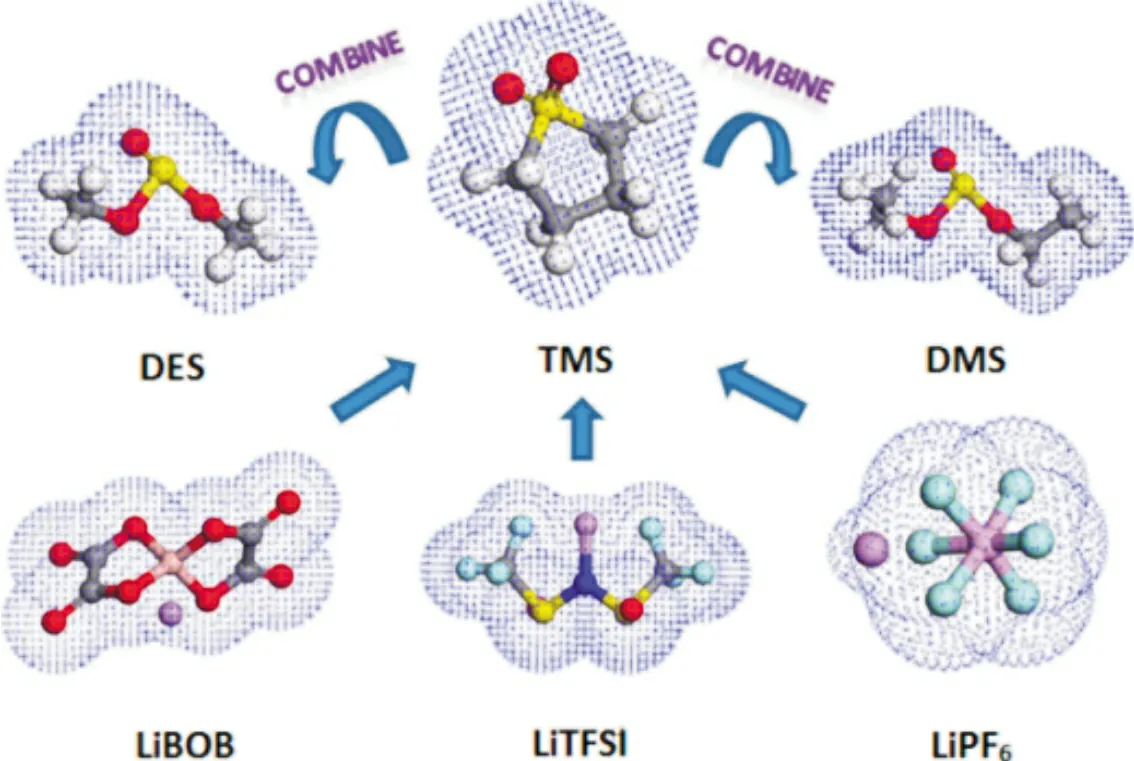
图3 锂盐和砜类溶剂的电子云结构[21]
高电压溶剂不仅有砜类溶剂和氟代溶剂还有腈类溶剂及离子液体等,但是他们各有优缺点,例如砜类溶剂及腈类溶剂粘度大,与负极兼容性差,氟代溶剂制作成本及安全要求高。为了趋利避害,研究者往往采用多种溶剂混合的方式来达到更优的效果,Kong 等[25]使用碳酸二甲酯/氟乙烯二丙醚/氟代碳酸乙烯酯/二甲基砜作为新型混合溶剂,与常规电解质相比,LCO 电池(vs.Li/Li+)在4.45 V 下循环300 次后,容量保持率从原来的38.1%提高到87.1%,在4.55 V 的更高电压下300 次循环后容量保持率也从以前的21.0%有效地提高到74.9%,并且该电解质与石墨负极保持很好的相容性。
2.2 成膜添加剂
相较于在正极材料上掺杂和包覆无机化合物来抑制钴离子溶出,在电解质中添加成膜添加剂是简单有效且成本低廉的方法。根据前线分子轨道理论,正极成膜添加剂分子的氧化电位需低于电解质溶剂,HOMO 能级应高于溶剂分子。
研究发现,一些含S 的成膜添加剂分子HOMO 能量高于常见的碳酸酯溶剂,这表明它们的氧化电位较低,在充电过程中优先在正极上氧化聚合(形成Li2SO3或ROSO2Li),形成一层保护膜抑制钴离子溶出及界面反应[26]。Zheng 等[27]在4.5 V的LCO 电池电解质(LiPF6,EC,DMC 和EMC)中加入0.5%二(甲基磺酰基)乙烷(DMSE)进行评估;在100 次充放电后,容量保持率从20.8%提高到66.5%。与碳酸酯溶剂相比,DMSE 具有更高的HOMO 能级和更低的氧化电位,由于较低的氧化稳定性,DMSE 会在LCO 表面上优先于溶剂分解形成CEI层,从而保护正极材料结构,抑制电解质溶剂进一步分解。在各种含S 添加剂中,噻吩类衍生物可以在电化学环境下聚合成聚噻吩,表现出高电导率和优异的化学稳定性。Sun 等[28]通过在基础电解液中添加0.5% 2-(三氟乙酰基)噻吩(TFPN),LCO电池的循环(0.5C,3.0~4.4 V,100 次,vs.Li/ Li+)容量保持率从33.2%提升至90.6%。
与其他成膜添加剂不一样的是腈类添加剂还可以与过渡金属(Co4+或Ni4+)络合,形成强配位键[R-CNδ-Metal(4+δ)+],防止过渡金属对电解质催化氧化,从而改善电池的循环寿命及热稳定性。Yang 等[29]通过在传统碳酸酯类电解质中添加亚甲腈(SUN)或1,3,6-己三腈(HTCN)可以实现LCO 电池(vs.Li/Li+)在4.6 V 截止电压下,300 次循环(30 ℃,1C)后容量保持率超过72%,200 次循环(55 ℃,1C)后容量保持率超过60%。这些改善得益于其在LCO 正极上形成超薄且均匀的CEI 膜,并抑制了微观结构的演变(Co4+孤对电子对与-CN 的N 2p 轨道的配位),可以有效降低Co4+的高价态,从而降低了过渡金属离子对整体电解质的催化反应性。
除此之外,还有含硼类添加剂[双草酸硼酸锂(LiBOB),草酸硼酸锂(LiDFOB)和三(三甲基硅基)硼酸酯(TMSB)等]不仅可以优先在正极被氧化参与修饰CEI 层,还可防止PF6-分解为PF5、LiF 或其他副产物,亲电子的含硼化合物也可以增加LiF 的溶解度,防止界面阻抗增加[30]。此外,硼酸盐化合物可以被认为是阴离子受体,它们与锂盐中的阴离子络合,不仅可以提高电解质中锂离子的导电性,而且可以提高锂离子的迁移数。
3 结论
掺杂元素的离子半径比钴离子大,因此它们可以在锂离子嵌入/脱出过程中稳定层状结构(立柱效应)。尽管对掺杂方法的研究已经进行了很长时间,但仍存在许多未知领域需要研究,例如F-的影响,多元素掺杂的影响以及抑制相变的机理等。包覆法可以构建LCO 材料保护层,以防止电解质对其腐蚀,并且与元素掺杂方法不同的是涂层使相变可逆而不是抑制相变。但是包覆法的难点在于涂层材料的锂离子迁移率。就液体电解质而言,多采用前线分子轨道理论计算与电化学实验相结合的方法探究其性能,电解质溶剂需要有更宽的HOMO 及LUMO“能带隙”,才能保证在高电压下溶剂整体性能的稳定,电解质添加剂则需要比溶剂高的HOMO 能级,以便在正极形成CEI。
