苯磺酸贝他斯汀的合成新方法
2022-03-16胡鑫欣赖月琴王浩宇张世明袁伟成周鸣强
胡鑫欣, 赖月琴, 王浩宇, 陈 宇, 张世明*, 袁伟成*, 周鸣强*
(1.遵义医科大学 药学院 贵州省生物催化与手性药物合成重点实验室,贵州 遵义 563000; 2.成都丽凯手性技术有限公司 手性药物国家工程研究中心,四川 成都 610000; 3.浙江金华康恩贝生物制药有限公司,浙江 金华 321016)
苯磺酸贝他斯汀(Chart 1)化学名为[S]-4-[(4-氯苯基)(2-吡啶基)甲氧基]-哌啶基]-丁酸苯磺酸盐,是日本田边公司和宇部公司联合开发的组胺H1受体拮抗剂,于2000年在日本获得批准上市,商品名为坦亮(TALION)。苯磺酸贝他斯汀具有血小板活化因子(PAF)拮抗作用,可抑制嗜酸粒细胞浸润至炎症组织,并可抑制白细胞介素-5(IL-5)、白三烯B4(LTB4)和血小板活化因子(PAF),减轻过敏炎症[1],常用于治疗过敏性鼻炎、荨麻疹、湿疹等。
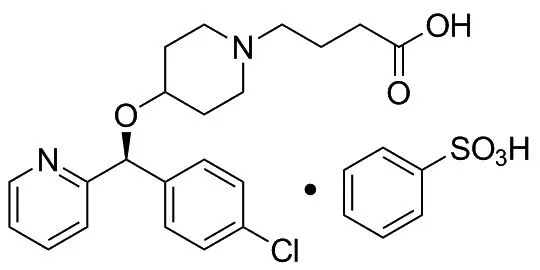
Chart 1
随着工业化进程的深入,空气污染指数大幅提高,过敏性炎症发病率逐渐增高[3]。临床应用证实,苯磺酸贝他斯汀具有起效快,嗜睡等不良反应发生率低等优势[4]。鉴于苯磺酸贝他斯汀的显著疗效以及广泛使用,该药物的高效合成引起药物合成工作者的高度兴趣。
目前,关于苯磺酸贝他斯汀的合成[5-8]主要涉及4条路线(Scheme 1)。综合分析这4条合成路线,发现存在以下不足之处:路线一[9]使用甲磺酰氯、N,N-二异丙基乙胺(DIPEA)等有毒溶剂,对人体有害;路线三、四使用NaH等强碱,安全隐患较大。路线二[11]需要130 ℃高温,反应条件苛刻,能耗高,副反应多。路线三[12]、四[13]由于贵金属催化剂的使用导致合成路线成本高,不利于工业化生产。
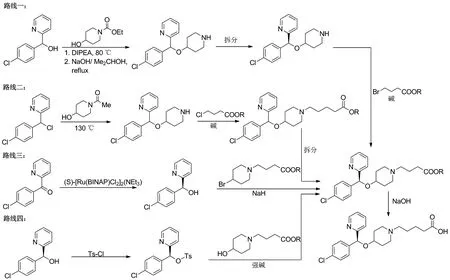
Scheme 1
通过分析已有合成路线,本文设计并开发了一种合成光学纯苯磺酸贝他斯汀的新方法(Scheme 2):以廉价易得的2-吡啶甲酸和氯苯为原料,通过傅-克反应、还原、酯化、醚化、拆分、缩合、水解,最后与苯磺酸成盐,得到光学纯苯磺酸贝他斯汀。其中,拆分副产物(R)-(4-氯苯基)(2-吡啶基)甲氧基哌啶可以经过消旋化后再次拆分得到(S)-构型产物进行利用。此合成方法中所涉及的原料和试剂廉价易得,合成方法更简单,条件温和,副反应少,具有可工业化生产前景。

Scheme 2
1 实验部分
1.1 仪器与试剂
Büchi S-545型熔点仪;Polarimeter 341型自动旋光仪;Bruker Avance-300 MHz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);Bruker Q TOF型质谱仪;Shimadzu LC20型高效液相色谱仪。
所用试剂均为分析纯或化学纯;试剂均未经进一步处理。
1.2 合成
(1)(4-氯苯基)(2-吡啶基)-甲酮(3)的合成
在反应瓶中加入2-吡啶甲酸(2.46 g, 20 mmol)和氯化亚砜10 mL,于80 ℃回流反应3 h。反应完毕后,减压浓缩至干。在冰浴冷却下往反应瓶中加入氯苯20 mL,搅拌下缓慢加入三氯化铝7.24 g(54 mmol),搅拌0.5 h后加热至108 ℃继续反应2 h(TLC检测)。反应完毕后,减压浓缩至干,0~5 ℃下缓慢滴加10%稀盐酸,并用二氯甲烷(30 mL)萃取,有机相用水(2×30 mL)洗涤,用无水硫酸钠干燥,减压浓缩至干。加入正己烷60 mL和活性炭400 mg回流脱色1 h,过滤,滤液冷却至室温,结晶得1.17 g白色粉末状固体3,收率30%, m.p.71.8~72.2 ℃;1H NMR(300 MHz, CDCl3)δ: 8.75~8.65(m, 1H), 8.06(d,J=8.6 Hz, 3H), 7.90(td,J=7.8 Hz, 1.7 Hz, 1H), 7.54~7.36(m, 3H);13C NMR(75 MHz, CDCl3)δ: 192.30, 154.57, 148.42, 139.29, 137.11, 134.48, 132.41, 128.37, 126.33, 124.58, 77.40, 76.97, 76.55; HR-MS(ESI)m/z: Calcd for C12H9ClNO{[M+H]+}218.0367, found 218.0360。
(2)(4-氯苯基)(2-吡啶基)-甲醇(4)的合成
在反应瓶中加入3(2.02 g, 10 mmol)和甲醇10 mL,搅拌下缓慢加入硼氢化钠0.21 g(5.5 mmol)。反应混合液室温搅拌反应1 h(TLC检测)。用乙酸乙酯(3×15 mL)萃取,合并有机相,用无水硫酸钠干燥,减压浓缩至干,用正己烷重结晶,得1.86 g纯白色固体4,收率85%,m.p.88.0~88.6 ℃;1H NMR(300 MHz, CDCl3)δ: 8.54(d,J=4.5 Hz, 1H), 7.62(td,J=7.7 Hz, 1.7 Hz, 1H), 7.38~7.07(m, 6H), 5.72(s, 1H), 5.39(s, 1H);13C NMR(75 MHz, CDCl3)δ: 160.39, 147.89, 141.69, 136.92, 133.52, 128.65, 128.34, 122.56, 121.16, 77.41, 76.99, 76.57, 74.26; HR-MS(ESI)m/z: Calcd for C12H11ClNO{[M+H]+}220.0524, found 220.0520。
(3)(4-氯苯基)(2-吡啶基)甲基-2,2,2-三氯乙酰亚胺酯(5)的合成
在反应瓶中加入4(4.40 g, 20 mmol)和二氯甲烷34 mL,降温至0~5 ℃,加入氢氧化钾 1.12 g(20 mmol),再缓慢滴加三氯乙腈5.20 g(36 mmol),滴毕,保温反应0.5 h(TLC检测)。加水20 mL,搅拌,分液,二氯甲烷相用水(3×20 mL)洗涤,无水硫酸钠干燥,减压浓缩得7.01 g棕黄色油状液体5,收率96%,m.p.87.7~88.2 ℃;1H NMR(300 MHz, CDCl3)δ: 8.58(d,J=4.1 Hz, 1H), 8.50(s, 1H), 7.72(td,J=7.7 Hz, 1.7 Hz, 1H), 7.60(d,J=7.9 Hz, 1H), 7.46(d,J=8.4 Hz, 2H), 7.33(dd,J=14.3 Hz, 7.5 Hz, 2H), 7.25~7.15(m, 1H), 6.95(s, 1H), 3.71(s, 1H), 1.89(s, 1H), 1.24(t,J=7.0 Hz, 1H);13C NMR(75 MHz, CDCl3)δ: 160.84, 158.64, 149.36, 137.08, 137.00, 134.06, 129.07, 128.66, 128.38, 127.41, 122.83, 120.89, 119.97, 91.14, 81.34, 77.38, 76.96, 76.53; HR-MS(ESI)m/z: Calcd for C14H10Cl4N2NaO{[M+Na]+}384.9439, found 384.9426。
(4)4-[(4-氯苯基)-2吡啶基甲氧基]-1-哌啶甲酸乙酯(6)的合成
在反应瓶中依次加入N-乙氧羰基-4-羟基哌啶(6.20 g, 35.8 mmol)、二氯甲烷150 mL和5(10.0 g, 27.5 mmol),降温至-20 ℃,缓慢滴加三氟甲磺酸4.13 g(27.5 mmol),并控制反应温度<-15 ℃。反应1.5 h(TLC检测)后,反应液减压浓缩至干,加入乙酸乙酯100 mL搅拌0.5 h,过滤,得12.0 g白色固体6,收率83 %,m.p.187.1~187.5 ℃;1H NMR(300 MHz, CDCl3)δ: 8.49(d,J=4.2 Hz, 1H), 7.67(td,J=7.7 Hz, 1.7 Hz, 1H), 7.51(d,J=7.9 Hz, 1H), 7.35(d,J=8.4 Hz, 2H), 7.30~7.21(m, 2H), 7.15(dd,J=6.4 Hz, 5.0 Hz, 1H), 5.60(s, 1H), 4.10(q,J=7.1 Hz, 2H), 3.74(s, 2H), 3.67~3.55(m, 1H), 3.17(qd,J=8.2 Hz, 4.0 Hz, 2H), 1.81(d,J=6.3 Hz, 2H), 1.72~1.54(m, 2H), 1.27~1.17(m, 3H);13C NMR(75 MHz, CDCl3)δ: 161.71, 155.40, 148.83, 139.98, 136.87, 133.24, 128.46, 128.01, 122.45, 120.45, 80.86, 77.38, 76.96, 76.54, 72.45, 61.16, 40.93, 40.90, 30.93, 14.58; HR-MS(ESI)m/z: Calcd for C20H23ClN2NaO3{[M+Na]+}397.1289, found 397.1280。
(5)(4-氯苯基)(2-吡啶基)甲氧基哌啶(7)的合成
在反应瓶中依次加入6(8.2 g, 15.7mmol)乙醇200 mL和氢氧化钾22.0 g,加热回流反应17 h(TLC检测)。反应液减压浓缩至干,加入水70 mL,用二氯甲烷(3×70 mL)萃取,合并有机相,依次用饱和食盐水(3×70 mL)洗涤,无水硫酸钠干燥,减压浓缩至干,得4.68 g棕黄色油状物7,收率98%,m.p.65.1~65.5 ℃;1H NMR(300 MHz, CDCl3)δ: 8.54~8.44(m, 1H), 7.66(td,J=7.7 Hz, 1.6 Hz, 1H), 7.52(d,J=7.9 Hz, 1H), 7.36(d,J=8.4 Hz, 2H), 7.32~7.19(m, 2H), 7.19~7.07(m, 1H), 5.62(s, 1H), 3.60~3.40(m, 1H), 3.06(dt,J=12.3 Hz, 4.2 Hz, 2H), 2.55(tt,J=13.0 Hz, 3.1 Hz, 2H), 1.90(dd,J=12.6 Hz, 8.9 Hz, 2H), 1.78(s, 1H), 1.68~1.42(m, 2H);13C NMR(75 MHz, CDCl3)δ: 162.01, 148.78, 140.29, 136.78, 133.11, 128.39, 128.09, 122.32, 120.52, 80.54, 77.38, 76.95, 76.53, 73.74, 44.20, 32.96, 32.92; HR-MS(ESI)m/z: Calcd for C17H20ClN2O{[M+H]+}303.1259, found 303.1247。
(6)(S)-(4-氯苯基)(2-吡啶基)甲氧基哌啶(8)的合成

(7)(S)-贝他斯汀乙酯(10)的合成
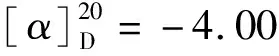
(8)(S)-贝他斯汀(11)的合成

(9)苯磺酸贝他斯汀(1)的合成
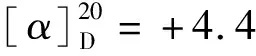
(10)(R)-(4-氯苯基)(2-吡啶基)甲氧基哌啶(9)消旋化
在反应瓶中依次加入富含9的拆分副产物(10.0 g, 33 mmol)、异丙醇200 mL和叔丁醇钾37.0 g,加热至回流反应17 h,降至室温过滤,滤液减压浓缩至干,加入二氯甲烷150 mL,有机相用水(3×75 mL)洗涤,无水硫酸钠干燥,减压浓缩至干,得泡沫状固体消旋体7。
在合成化合物5的过程中,筛选了KOH、t-BuOK、DBU、NaH、NaOH等碱性催化剂,其中t-BuOK反应杂质较多且产率低,DBU为液体溶于有机相不易除去,导致操作困难。采用KOH为催化剂,收率最高,且KOH作为固体碱,易去除,后处理简单,成本低,适合工业化生产操作。在合成化合物6的过程中,重点考虑了5和N-乙氧羰基-4-羟基的投料比,发现两者比例为1/1.3时收率明显增加,继续提高比例,收率基本维持不变。合成化合物7主要涉及到脱N-乙氧羰基,我们也尝试了各种酸或者碱体系,如CH3COOH/HCl, CH3COOH/HBr, IPA/KOH, CH3CH2OH/KOH,发现CH3CH2OH/KOH体系反应效果最好,而CH3COOH/HCl体系则无法反应。最后,从化合物7到合成化合物8,主要涉及拆分,我们也尝试了各种拆分剂,具体结果如(表1)。从表1可以看出,L-DTTA、N-乙酰-L-亮氨酸、L-DBTA、L-酒石酸、D-DBTA有一定效果,其中D-DBTA拆分效果较好。

表1 (4-氯苯基)(2-吡啶基)甲氧基哌啶的拆分
开发了一条合成苯磺酸贝他斯汀的新路线。以2-吡啶甲酸、氯苯为原料,经过9步反应得到光学纯的苯磺酸贝他斯汀。利用D-DBTA拆分关键中间体(4-氯苯基)(2-吡啶基)甲氧基哌啶,得到光学纯的(S)-异构体(28% yield, 99.8%ee)。同时可以将R-(4-氯苯基)(2-吡啶基)甲氧基哌啶消旋化处理,回收利用,大大提高产率并降低生产成本。该合成路线所涉及的原料廉价易得、合成方法简单、条件温和、副反应少,具有较好的工业化生产前景。
