2015-2017年新生儿原发性肉碱吸收障碍串联质谱法回顾性分析*
2022-02-19程昱璇张春燕崔佳奕檀旭东桑培培史文杰张民杰田亚平
程昱璇,张春燕,舒 杨,蒋 涛,崔佳奕,檀旭东,桑培培,史文杰,张民杰,田亚平
中国人民解放军总医院医学创新研究部出生缺陷防控技术研究中心,北京100853
原发性肉碱吸收障碍(CUD)又称原发性肉碱缺乏症或肉碱转运障碍,是常染色体隐性遗传的脂肪酸β氧化代谢病,是一种与细胞线粒体有关的疾病[1]。CUD是由于SLC22A5基因(位于染色体5q31.1上,约30 kb)突变导致细胞膜有机阳离子转运体2(OCTN2)缺陷,造成多个组织受累包括骨骼肌、心肌、脑和肾脏等组织内肉碱水平不足以支持长链脂肪酸进入线粒体参与氧化[2]。CUD发生率约为0.8/10万~2.5/10万,不同地区发生率存在差异,其为脂肪酸氧化代谢的常见疾病[3]。
本研究采用血液串联质谱(MSMS)筛查的方法,对10.9万份各地标本进行初筛,现对这部分标本的肉碱检测结果作回顾性分析报道。
1 资料与方法
1.1一般资料 选择2015-2017年于本院出生缺陷防控技术研究中心进行新生儿MSMS筛查的10.9万份滤纸干血片标本,在新生儿父母签署知情同意书的前提下,用 MSMS进行多种氨基酸、肉碱和琥珀酰丙酮的检测分析,标本来源于四川、新疆、西藏、贵州、湖北、北京出生的新生儿。
1.2仪器与试剂 使用美国Waters Xevo TQD质谱仪、美国Waters 2777自动进样系统、1525u高效液相仪、孵浴震荡仪、美国 PerkinElmer 流动相和萃取溶剂试剂盒(含甲醇、水和草酸)、美国 PerkinElmer非衍生化串联质谱试剂盒、琥珀酰丙酮内标液进行前处理并上机检测。
1.3方法
1.3.1MSMS试验分析 用包含稳定同位素标记的氨基酸、酰基肉碱和琥珀酰丙酮的内标准品的溶剂萃取滤纸干血片中的氨基酸、游离肉碱(C0)和酰基肉碱,然后用MSMS系统分析。具体操作步骤如下:用1 mL萃取液溶解内标干粉制成储备工作液,于2~8 ℃保存待用。用直径3 mm打孔钳打取滤纸片至96孔板中,按照萃取液与内标液110∶1的比例配制萃取工作液,每孔加入含氨基酸和酰基肉碱同位素内标工作液100 μL,45 ℃,700 r/min密封孵育震荡45 min,提取80 μL萃取液转移至V型底的96孔板内,铝膜覆盖,放置在自动进样器上静置2 h后即可检测[4]。采用美国Waters公司Masslynx软件处理数据,由已知水平的内标自动计算出所测样品的氨基酸、酰基肉碱和琥珀酸丙酮的水平。同一标本异常指标2次结果均超过参考值,判为筛查阳性者立即召回。对C0偏低的患儿母亲同时进行血液酰基肉碱水平分析。
1.3.2SLC22A5基因测序分析 筛查出新生儿阳性标本(C0水平低于9.5 μmol/L),采用干血片法提取基因组DNA,进行全外显子测序(WES),测序结果与人类基因组SLC22A5基因序列进行比较[5]。
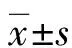
2 结 果
在10.9万份筛查标本中初次筛查提示CUD(或继发性肉碱吸收障碍)124例,其中男54例、女70例,平均年龄(164.40±16.18)d,平均体质量(3 346.00±54.49)g,平均C0为(6.27±0.16)μmol/L,本实验室C0的参考范围为9.5~48.0 μmol/L。共召回39例患儿标本再次进行二次筛查,最终对确定6例疑似患儿进行基因确诊,其中3例提示基因突变,对应C0及酰基肉碱谱水平见表1。3例患儿SLC22A5基因诊断到2种突变,2例c.1400C>G(p.S467C)杂合突变和1例c.51C>G(p.F17L)纯合突变,见表2。病例1患儿因丙二酰基肉碱水平升高召回,基因检测发现另有SLC22A5基因c.1400C>G(p.S467C)杂合突变,C0水平为12.37 μmol/L。病例2患儿C0水平为4.14 μmol/L,召回基因检测发现SLC22A5基因c.1400C>G(p.S467C)杂合突变,肉碱及酰基肉碱谱整体降低。病例3患儿C0水平为3.57 μmol/L,召回基因检测发现SLC22A5基因c.51C>G(p.F17L)纯合突变,肉碱及酰基肉碱谱整体降低明显。

表1 3例CUD患儿C0及酰基肉碱谱水平(μmol/L)
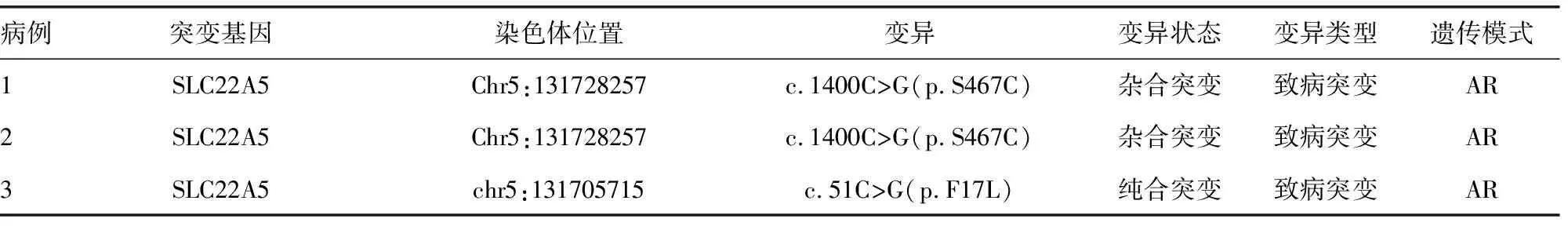
表2 3例CUD患儿基因检测结果
3 讨 论
研究表明在法罗群岛CUD的患病率最高为1∶300[6],日本的患病率约为1∶40 000[7],澳大利亚为1∶37 000~1:100 000[8],美国为1∶142 000[9],在中国河南省的患病率为1∶31 499[10],浙江省为1∶23 862[11],上海市为1∶45000[1],河北省为1∶17 785[12],本研究共筛查来自四川、新疆、西藏、贵州、湖北、北京6地新生儿标本10.9万例,通过MSMS筛查出124例疑似阳性标本,阳性疑似率约为1.14‰,确诊CUD 3例,患病率约为1∶36 333,与报道的中国CUD新生儿筛查患病率相符[12]。MSMS技术对早期发现CUD有提示意义,但最终确诊需结合其他检测手段,如尿有机酸检测和基因检测等,并结合临床明确诊断。
CUD是由SLC22A5基因编码的OCTN2肉碱转运蛋白突变引起的常染色体隐性(AR)遗传疾病,有10个外显子[13]。SLC22A5突变数据库(http://www.arup.utah.edu/database/OCTN2/OCTN2_display.php)中描述了150多种遗传变异,其中86种具有致病性(包括错义、无义、插入缺失、移码,剪接位点改变突变和一个大基因缺失)[13]。大多数研究认为c.760C> T(p.R254X)和c.1400C>G(p.S467C)是中国人最常报道的突变[14]。本研究结果检测出2例c.1400C> G(p.S467C)和1例c.51C>G(p.F17L),c.1400C>G(p.S467C)占比2/3,与报道相一致[15]。基因突变致其编码的肉碱转运蛋白无法锚定细胞膜而滞留于细胞质中,或结构及功能域不同程度受损,导致该蛋白的转运功能缺陷。肉碱(β-羟基-γ-三甲基丁酸铵)是一种衍生自氨基酸的必不可少的水溶性分子[15]。在非素食主义者中,肉碱的主要来源是红肉,家禽和奶制品[16],几乎占人体总贮存量的3/4[17]。肉碱由肠道内转入到血液及由血液内转入到细胞的量减少,血液及细胞内肉碱缺乏,尿流失增加,脂肪酸β氧化代谢受阻[18]。CUD可于任何年龄发病,患者表现为心肌疾病、心功能降低、肌无力、肌张力减退及肝功能异常等[19],若是不及时医治,可导致死亡。有些患儿在诊断时通常是完全无症状的,可通过测量血浆肉碱水平(游离和总量)和研究肉碱在成纤维细胞中的转运来确认[15]。左卡尼汀的治疗,使越来越多CUD患儿的预后得到了改善[20-21]。CUD是为数不多的治疗效果良好的遗传病之一,所以早期诊断、及时治疗是改善患儿预后的关键。
本研究通过足跟采血,滴于专用滤纸片后晾干,寄送到本实验室使用MSMS方法测定C0及其他酰基肉碱,CUD患儿C0及多种酰基肉碱水平降低,C0水平正常参考值为9.5~48.0 μmol/L。病例1患儿因丙二酰基肉碱水平升高召回,基因检测发现另有SLC22A5基因c.1400C>G(p.S467C)杂合突变,C0串联检测结果正常。病例2患儿C0水平为4.14 μmol/L,召回基因检测发现SLC22A5基因c.1400C>G(p.S467C)杂合突变,肉碱及酰基肉碱谱整体降低。病例3患儿C0水平为3.57 μmol/L召回基因检测发现SLC22A5基因c.51C>G(p.F17L)纯合突变,肉碱及酰基肉碱谱整体降低明显。杂合突变个体的肉碱转运活性为正常个体的50%,C0值可处于临界低水平[22]。此外,C0能通过胎盘从母体转运给胎儿,若母亲体内肉碱充足,CUD胎儿可以从母体获得肉碱供给,在生后的一段时间内仍保持较充足的肉碱储备,导致筛查时出现假阴性。若母亲为原发性肉碱缺乏症或各种原因导致血液中肉碱不足,也会导致新生儿筛查时C0水平低于正常[23],导致假阳性,故新生儿筛查阳性者,需要同时测母亲C0水平,以便除外因母体原因导致的继发性肉碱缺乏。
CUD是一种潜在的致死性疾病,早期诊断和治疗是决定预后的关键。本研究使用MSMS技术筛查10.9万份标本,检测出124例CUD疑似阳性患儿,对可疑阳性患者的标本进行SLC22A5基因测序,进一步确诊了3例CUD患儿。MSMSM技术的应用使CUD的早期诊断成为可能,结合测序技术也为CUD 的遗传咨询和产前诊断提供了重要信息。
