HPLC测定羟氯扎胺阿苯达唑复方混悬液的含量及有关物质
2021-12-24张东辉白玉彬申涵露周绪正
张东辉,白玉彬,董 朕,陈 晨,申涵露,李 冰,周绪正
(中国农业科学院 兰州畜牧与兽药研究所 农业部兽用药物创制重点实验室 甘肃省新兽药工程重点实验室,甘肃 兰州 730050)
羟氯扎胺(oxyclozanide,OXY)又名五氯柳胺,是水杨酰苯胺类抗寄生虫药物。作为氧化磷酸化解偶联剂,其主要作用机理是通过抑制寄生虫虫体内的氧化磷酸化过程,来阻止虫体线粒体中的三磷酸腺苷(ATP)生成,进而导致虫体因能量代谢紊乱而死亡[1]。OXY具有广谱、低毒、低残留等优良特性[2-4],主要用于治疗牛羊吸虫病,尤其对肝片吸虫治疗效果更为突出,同时对绦虫、线虫也有良好的治疗效果[5-7]。阿苯达唑(albendazole,ABZ)又名丙硫咪唑,是苯并咪唑类驱虫药,能选择性与不可逆性抑制虫体摄取葡萄糖,使虫体内源性糖原耗竭,并抑制延胡索酸酶系统,阻碍ATP的产生,致使虫体死亡,是一种广谱、高效、低毒的驱虫药[8]。
由于牧区养殖环境的复杂性,牛、羊等经济型动物在养殖过程中往往会出现多种寄生虫混合感染[9-10],在治疗过程中因可选用的复方制剂缺乏,只能选用多种单一驱虫药联合给药,或者选用广谱驱虫药进行治疗。目前国外的复方制剂有羟氯扎胺盐酸左旋咪唑混悬液,而国内复方制剂只有阿苯达唑伊维菌素预混剂,因此研发一种新的抗寄生虫复方制剂事不宜迟。本团队研发的羟氯扎胺阿苯达唑复方制剂可以起到增加抗虫谱,减少给药剂量等作用,避免单一驱虫药频繁使用而产生耐药性问题,有效的促进畜牧业健康发展。李晓婷等[11]采用HPLC只对OXY原料药中的2个杂质进行检测,为了更严格地控制复方混悬液的质量,本试验将采用高效液相色谱法建立了羟氯扎胺阿苯达唑复方混悬液的含量及有关物质测定方法,对OXY、ABZ在2015版《英国药典》中规定的所有杂质进行测定[12-14]。OXY、ABZ及各相关杂质化学结构式见图1。

图1 ABZ及相关杂质A~F和OXY及相关杂质G、H的化学结构式
1 材料与方法
1.1 主要仪器Agilent-LC 1290高效液相色谱仪(安捷伦科技有限公司);ME403型电子天平(梅特勒-托利多仪器有限公司);KQ-600DE数控超声波清洗器(昆山市超声仪器有限公司);ΜPT-Ⅱ-40L优普超纯水制造系统(上海优普实业有限公司)。
1.2 主要试剂OXY标准品(含量质量分数:99.7%,批号:RSB-160703,Dr.Ehrenstorfer GmbH公司);ABZ标准品(含量质量分数:99.9%,批号:100373-201103,中国食品药品检定研究院);3,5,6-三氯水杨酸标准品(含量质量分数:99.0%,批号:151708,阿拉丁试剂有限公司);2-氨基-4,6-二氯苯酚盐酸盐(含量质量分数:98.0%,批号:A2246,英国药典委员会);ABZ杂质A、B、C、D、E、F对照品(含量质量分数分别为98.0%,98.0%,98.0%,98.0%,98.0%和97.0%,Trc公司);羟氯扎胺阿苯达唑复方混悬液(含量:4.0%,中国农业科学院兰州畜牧与兽药研究所研制)。
甲醇、冰乙酸(分析纯,国药集团化学试剂有限公司);甲醇(色谱纯,Merck);甲酸、甲酸铵(色谱纯,Flsher);超纯水(优普)。
1.3 色谱条件色谱柱:Agilent Poroshell EC-C18(150 mm×4.6 mm,2.7 μm);柱温:35℃;进样量:3 μL;流速:0.8 mL/min;流动相:A相为0.1%甲酸-10 mmol/L甲酸铵水溶液,B相为甲醇;检测波长:292,320 nm;梯度洗脱如表1。

表1 HPLC梯度洗脱条件 %
1.4 溶液制备
1.4.1供试品溶液 取羟氯扎胺阿苯达唑复方混悬液适量(约相当于OXY和ABZ各80 mg),精密称定,置于100 mL量瓶中,加入适量冰乙酸、甲醇分别超声10 min,最后再用甲醇定容至刻度,摇匀,即为供试品溶液。
1.4.2对照品溶液 精密移取1.4.1供试品溶液1 mL,置于100 mL量瓶中,用流动相定容至刻度,充分摇匀,即为对照品溶液。
1.4.3空白辅料 按处方比例称取羟氯扎胺阿苯达唑复方混悬液辅料适量(约相当于OXY和ABZ各80 mg),置于100 mL量瓶中,用流动相稀释并定容至刻度,摇匀。
1.4.4对照品储备液 分别精密称取OXY、ABZ标准品约20 mg及杂质A、B、C、D、E、F对照品各约4 mg,3,5,6-三氯水杨酸约60 mg和2-氨基-4,6-二氯苯酚盐酸盐约6 mg置于20 mL量瓶中,用适量DMSO超声溶解,再用甲醇定容至刻度,作为对照品储备液Ⅰ。精密移取对照品储备液Ⅰ1.0 mL,置于20 mL量瓶中,用甲醇定容至刻度,摇匀,作为对照品储备液Ⅱ。
1.4.5系统适用性溶液 精密称取OXY和ABZ标准品约1 mg,置于50 mL量瓶中,用冰乙酸酸化的甲醇溶解。精密量取对照品储备液Ⅱ适量,用甲醇定容至刻度,摇匀,最终配制成主成分与杂质比例为200∶10的专属性溶液,即为系统适用性溶液。
1.5 专属性试验分别取1.4.1供试品溶液、1.4.3空白辅料溶液和1.4.5系统适用性溶液,按1.3色谱条件进样,记录色谱图。
1.6 破坏试验取羟氯扎胺阿苯达唑复方混悬液适量(约相当于OXY、ABZ各80 mg)5份,置于100 mL量瓶中,加入适量冰乙酸、甲醇各超声10 min,分别经酸、碱、氧化、光照、高温破坏后,加甲醇稀释至刻度,摇匀,作为破坏溶液。破坏试验操作如下:
强酸破坏:加入1 mol/L盐酸溶液4.0 mL,室温条件下放置2 d后,加入1 mol/L氢氧化钠溶液4.0 mL中和;强碱破坏:加入1 mol/L氢氧化钠溶液4.0 mL,室温条件下放置2 d后,加入1 mol/L盐酸溶液4.0 mL中和;氧化破坏:加入体积分数为3%双氧水4.0 mL室温条件下避光放置30 min;光照破坏:置于光照箱内(4 500±500) lx照射2 d;高温破坏:置于药物稳定仪中,高温60℃,放置2 d。
分别吸取上述破坏试验样品溶液3 μL,按1.3色谱条件进样分析,记录色谱图。
1.7 精密度进样精密度:精密量取1.4.5系统适用性溶液3 μL,按1.3色谱条件进样分析,连续分析6次,计算各个杂质与主成分的保留时间和峰面积的RSD。
中间精密度:精密量取1.4.1供试品溶液6份,由甲、乙2人分别在不同时间段,使用2台不同仪器,按1.3色谱条件进样分析,计算各个杂质与主成分的保留时间和含量的RSD。
1.8 定量限与检测限取1.4.4各杂质对照品及ABZ、OXY主成分对照品储备液Ⅰ逐级稀释,按1.3色谱条件进样分析,记录色谱图。以信噪比S/N为3时对应的浓度为检测限(LOD);以信噪比S/N为10时对应的浓度为定量限(LOQ)。
1.9 线性范围考察分别精密移取1.4.4下ABZ、OXY以及各杂质对照品储备液Ⅱ0.10,0.25,0.40,0.50,0.60 mL于5 mL量瓶中,用甲醇定容至刻度,摇匀,即得到不同浓度的线性标准溶液。精密量取3 μL,按1.3色谱条件进样分析,记录色谱图。以峰面积(A)为纵坐标,质量浓度(ρ,mg/L)为横坐标,按最小二乘法进行线性回归。
1.10 重复性试验按1.4.1方法平行制备6份供试品溶液,按1.3色谱条件进样分析。
1.11 溶液稳定性按1.4.1和1.4.2方法制备供试品溶液和对照品溶液,在室温条件下放置,分别于0,2,4,6,8,12 h取样,按1.3色谱条件的进样分析,计算各杂质保留时间和峰面积的RSD值。
1.12 回收率试验分别精密称取OXY、ABZ对照品8 mg,置于10 mL量瓶中,共9份,按处方量称取空白辅料,加入适量冰乙酸、甲醇分别超声10 min,精密量取1.4.4杂质对照品储备液Ⅱ0.8,1.0,1.2 mL,并用甲醇定容至刻度,摇匀,作为低、中、高(80%,100%,120%)3个浓度水平的供试品溶液,每个水平平行制备3份。按1.3色谱条件进样分析,计算各回收率样品中杂质的含量(按外标法扣除本底值之后计算)。
1.13 耐用性试验取系统适用性溶液,在1.3色谱条件下,通过改变流速(±10%)、柱温(±5℃)、流动性比例(±2%),换用不同类型的不同厂家色谱柱,观察各组分之间的分离情况以及有关物质含量的变化。
1.14 有关物质含量测定取羟氯扎胺阿苯达唑复方混悬液3批样品(批号:20201007、20201008、20201009),按1.4方法制备供试品溶液和自身对照品溶液,按1.3色谱条件进样分析。采用不加校正因子的主成分自身对照法计算杂质含量,记录供试品溶液和自身对照品溶液色谱图至主成分保留时间的2倍,其中供试品溶液中任何小于自身对照品溶液主峰面积的0.05倍的峰可忽略不计。
2 结果
2.1 专属性试验由图2可知,各杂质与主成分OXY和ABZ之间以及各杂质之间的峰分离度均大于1.5,且峰形良好,出峰顺序依次为杂质D、杂质E、杂质B、杂质C、杂质F、杂质A、2-氨基-4,6-二氯苯酚盐酸盐、3,5,6-三氯水杨酸、ABZ和OXY,同时空白辅料不干扰测定,表明本方法专属性好。
2.2 破坏性试验由图3可知,羟氯扎胺阿苯达唑复方混悬液在强酸、强碱、光照、氧化的破坏条件下均有降解,主要降解产物是杂质B(阿苯达唑亚砜);在高温破坏后主要降解产物是杂质B(阿苯达唑亚砜)和杂质A(5-丙基硫烷基-1H-苯并咪唑-2-胺);在强光和氧化破坏中,主要是ABZ发生降解,其降解产物为杂质B。在各种破坏条件下,羟氯扎胺阿苯达唑主峰与各杂质峰均能良好分离,物料守恒均在90.0%~100.0%,分离度大于1.5,表明在本色谱条件下可以有效分离和检测各强制破坏降解产物,同时为各有关物质的来源做充分判断依据。
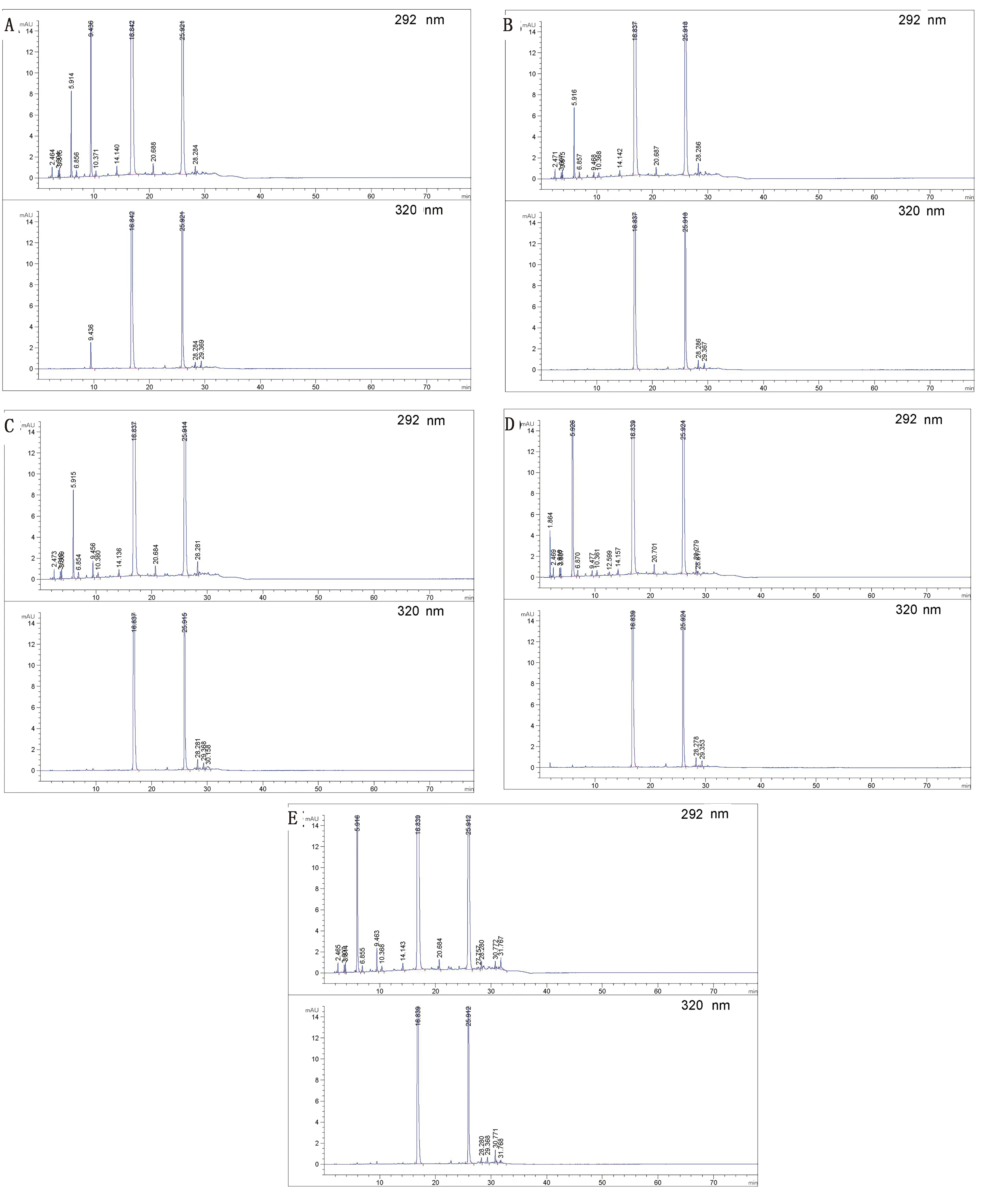
图3 高温(A)、酸(B)、碱(C)、氧化(D)和光照(E)破坏的色谱图
2.3 精密度
2.3.1进样精密度 连续进样6次,其各个杂质与主成分的保留时间和峰面积的RSD均小于2.0%,表明仪器精密度良好,仪器较稳定。
2.3.2中间精密度 分别由甲、乙2个人在不同时间,使用不同仪器连续进样分析6次,其各个杂质与主成分的保留时间和峰面积的RSD均小于2.0%,表明中间精密度良好。
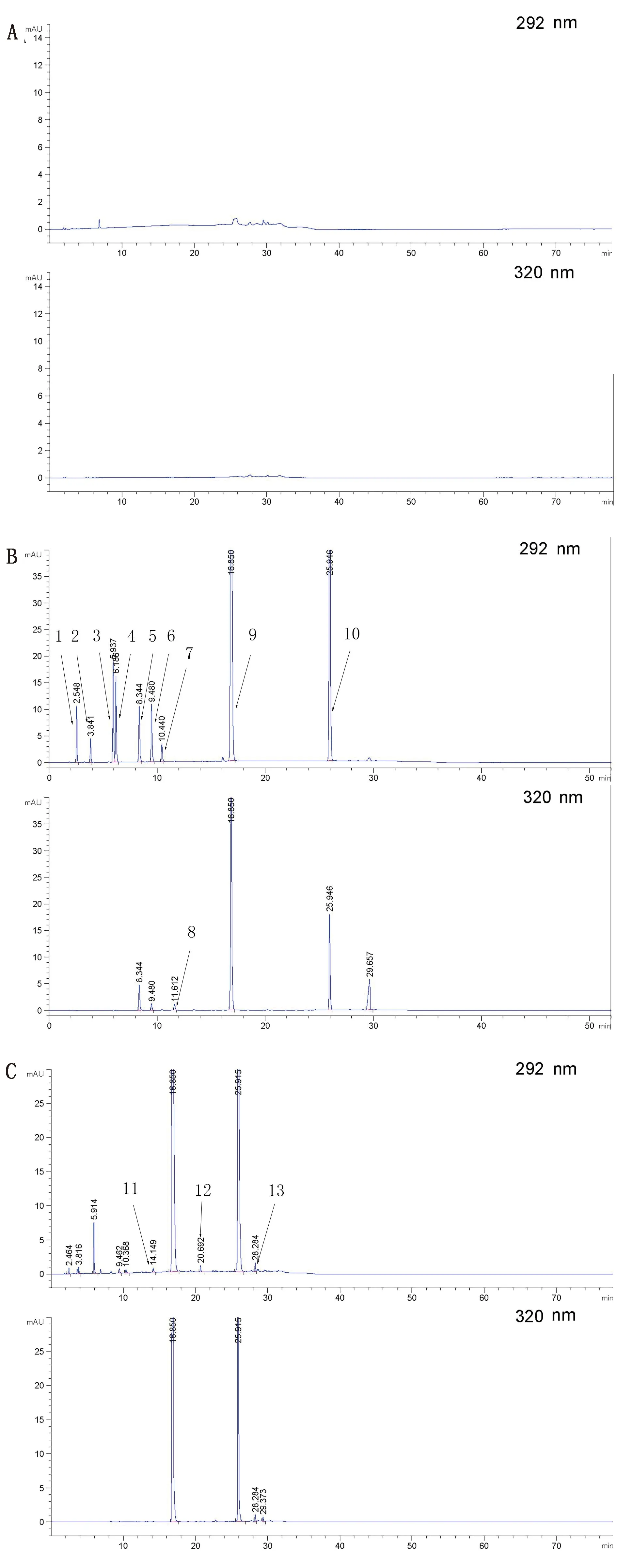
1.杂质D;2.杂质E;3.杂质B;4.杂质C;5.杂质F;6.杂质A;7.2-氨基-4,6-二氯苯酚盐酸盐;8.3,5,6-三氯水杨酸;9.ABZ;10.OXY;11.未知杂质1;12.未知杂质2;13.未知杂质3图2 空白辅料(A)、系统适用性溶液(B)和羟氯扎胺阿苯达唑复方混悬液(C)的色谱图
2.4 定量限与检测限试验结果表明,当S/N为10时,杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、3,5,6-三氯水杨酸、2-氨基-4,6-二氯苯酚盐酸盐、OXY、ABZ的定量限分别为0.10,0.06,0.07,0.10,0.15,0.13,5.00,0.30,0.10,0.30 mg/L;当S/N为3时,杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、3,5,6-三氯水杨酸、2-氨基-4,6-二氯苯酚盐酸盐、OXY、ABZ的检测限限分别为0.030,0.020,0.022,0.030,0.050,0.032,2.000,0.100,0.025,0.012 mg/L。
2.5 线性范围考察以峰面积(A)为纵坐标,质量浓度(ρ,mg/L)为横坐标进行线性回归,得到各个杂质与主成分的线性回归方程,其R2均大于0.999,表明线性关系良好。各个杂质与主成分的线性回归方程见表2。
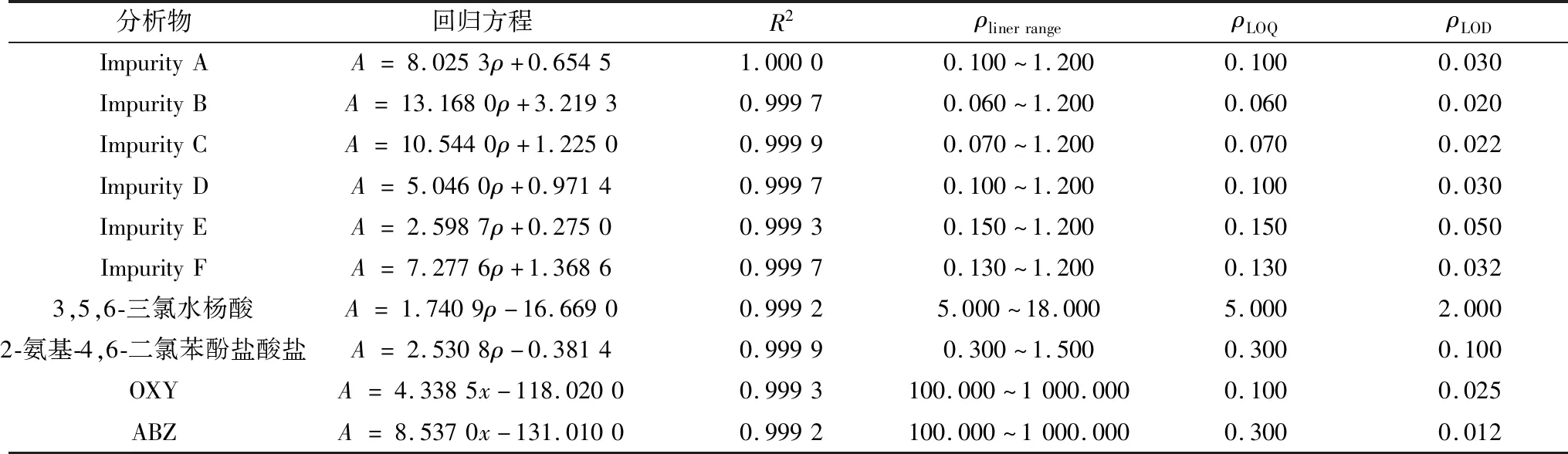
表2 OXY、ABZ及各个杂质的线性、定量限和检测限结果 mg/L
2.6 重复性试验结果显示,主成分OXY、ABZ以及各个杂质的保留时间和峰面积的RSD值均小于2.0%,表明该方法重现性良好,试验结果见表3。

表3 重复性试验结果 %
2.7 溶液稳定性在不同时间段对供试品溶液和对照品溶液取样分析,各个杂质保留时间与峰面积的RSD值均小于2.0%,且无新杂质产生,表明供试品溶液在12 h 内稳定,试验结果见表4。
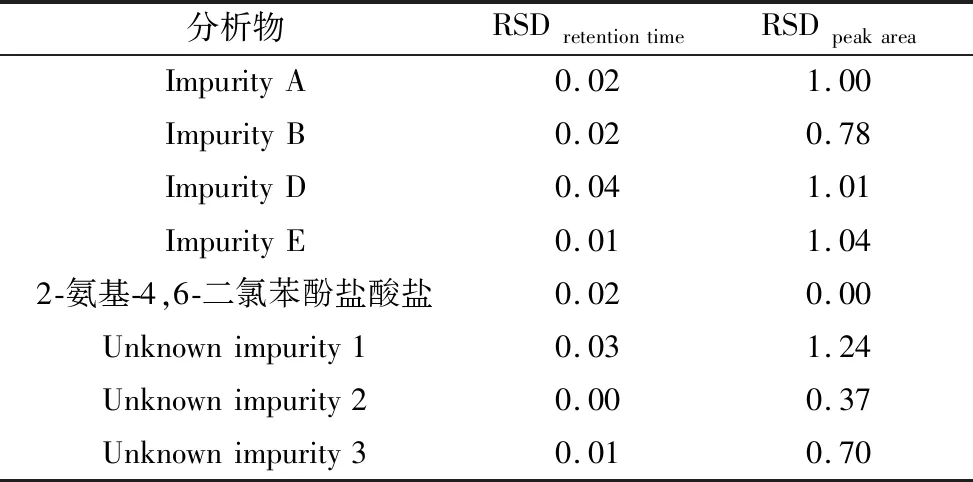
表4 溶液稳定性试验结果 %
2.8 回收率试验由表5可知,各个杂质及其主成分在低、中、高3个浓度下的回收率均在97.0%~107.0%。
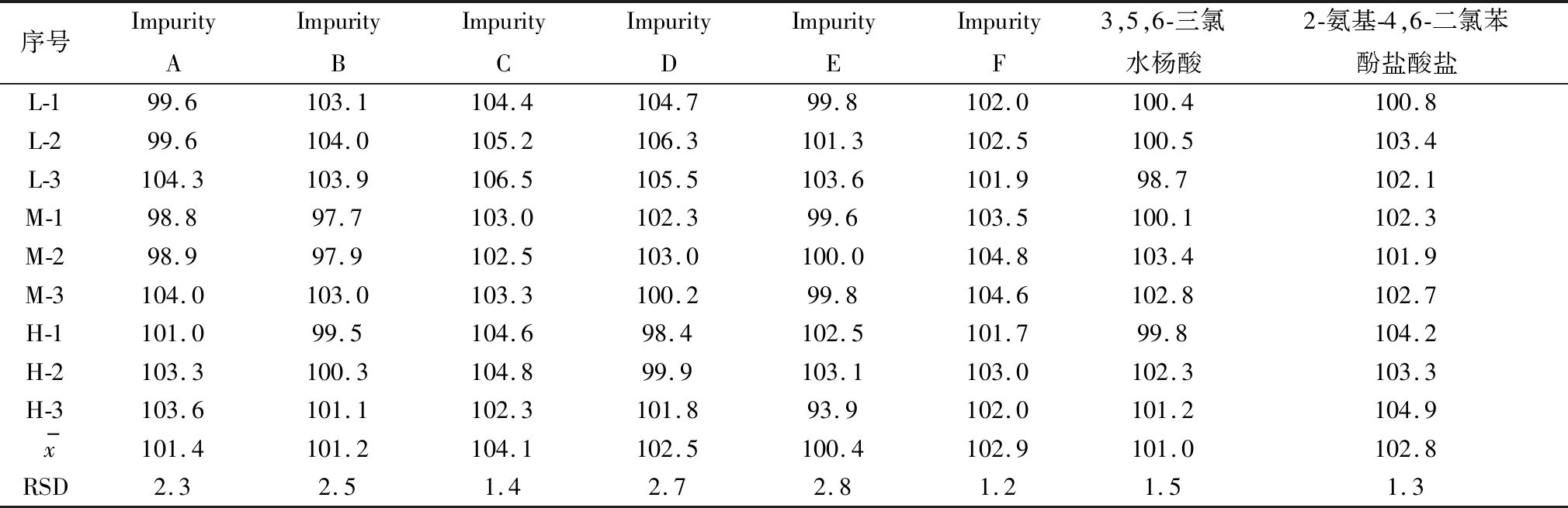
表5 加样回收率试验结果(n=9) %
2.9 耐用性试验结果表明,在各个条件下,主峰与各杂质峰均能分离,且各杂质含量的RSD均小于2.0%,结果说明在流动相比例、柱温、流速及色谱柱的微小变化对有关物质含量测定结果无显著性影响,表明该方法耐用性良好。
3.1 有关物质含量测定采用上述色谱方法对3批样品有关物质进行测定,由表6可知,样品中含有3个未知杂质,杂质B含量略高但均小于0.5%;所有单杂质含量均小于0.5%,总杂质含量均小于1.0%。
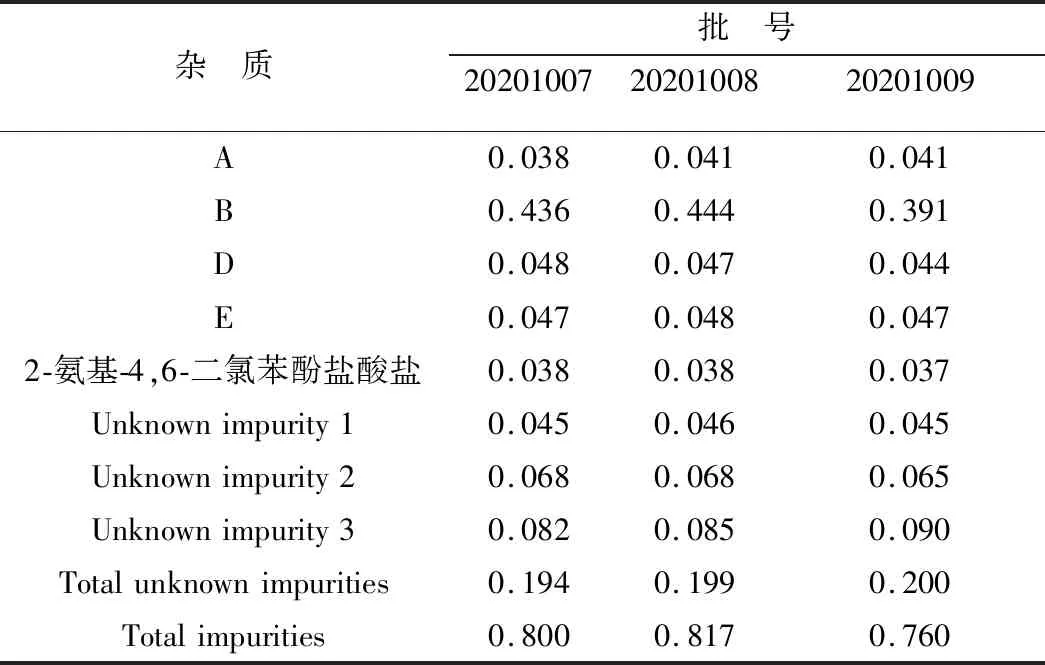
表6 样品有关物质的测定结果(n=3) %
3 讨论
3.1 检测波长分别取1.4各对照品溶液,用紫外分光光度计进行全波长扫描,杂质3,5,6-三氯水杨酸最大紫外吸收在320 nm处,其余各杂质在292 nm处均有最大吸收,这与英国药典中ABZ混悬液采用292 nm波长检测有关物质含量,所采用的波长相一致。同时由于复方制剂各成分之间比较复杂,杂质也相对较多,采用单波长很难充分检出[15-16],因此,采用292和320 nm双波长对羟氯扎胺阿苯达唑复方混悬液有关物质进行检测,灵敏度更高,准确性更好,能够准确定量检测。
3.2 色谱条件的确定根据相关文献[17-19],并参考各化合物的PKa值,本试验首先对甲醇-水、甲醇-磷酸盐和乙腈-磷酸盐3种梯度洗脱系统进行考察,结果发现采用乙腈-磷酸盐缓冲系统时,2-氨基-4,6-二氯苯酚盐酸盐与主成分ABZ分离效果较差,且ABZ和OXY主峰峰形分别出现前倾和拖尾现象。采用甲醇-磷酸盐缓冲系统时,在OXY主峰后出现未知峰,影响分离度,分别采用钠盐、钾盐、铵盐均无法消除。所以本试验确定以甲醇-甲酸铵缓冲体系作为流动相。其次为改善色谱峰峰形并提高分离度,对流动相添加剂如磷酸、三氟乙酸、甲酸等进行考察,结果显示0.1%甲酸峰形较好,且各杂质与主峰之间的分离效果最佳,故最终流动相选择甲醇-0.1%甲酸-10 mmol/L甲酸铵缓冲系统。
3.3 有关物质来源分析通常复方制剂在生产的过程中产生的杂质情况比较复杂,且据2013版《英国药典》《欧洲药典》8.0版收藏的ABZ原料药中有A、B、C、D、E、F 6个杂质,OXY原料药有2个杂质。对强酸、强碱、高温、氧化、光照等破坏试验产生的降解产物进行分析,ABZ、OXY在复方制剂中较为稳定,对酸、碱均不敏感,其有关物质的产生主要与ABZ氧化破坏有关,这与黄毅等[14]结果相一致,说明其主要杂质来源是药物的氧化产物,可能ABZ分子中含有丙硫基,易被氧化为杂质B(阿苯达唑亚砜),阿苯达唑亚砜是主要的降解产物,且阿苯达唑亚砜作为ABZ的活性物质,是体内发挥杀虫作用的主要成分,其本身不具有毒性,因此杂质B能间接反映样品的生产工艺、处方以及贮存条件变化情况,对羟氯扎胺阿苯达唑复方混悬液质量控制有着重要意义。在高温破坏性试验中表明未知杂质1主要是由高温产生,且在ABZ原料药中也检测到未知杂质1,因此在储藏过程中要阴凉处保存;白玉彬等[20]研究表明OXY混悬液有2个未知杂质,是OXY原料药在合成过程中产生的工艺杂质,用1.3色谱条件对OXY混悬液供试品进样分析,发现未知杂质2和3与OXY混悬液中的2个未知杂质保留时间一致,故该杂质可能来源于OXY原料药中。因此需要将已知杂质B和未知杂质1,2,3进行重点控制。
3.4 杂质限度的确定经检测3批样品中杂质B的含量分别为0.436%,0.444%,0.391%,未知杂质的总量为0.194%,0.199%,0.200%。在2013版《英国药典》对于ABZ采用HPLC法测定有关物质,规定单个杂质峰面积不得大于对照品溶液主峰面积的1.0%,各杂质峰面积之和不得大于对照品主峰面积的2倍(2.0%),而2015版《中国药典》对于ABZ有关物质测定依旧采用薄层色谱法;本团队在前期建立的HPLC检测OXY有关物质的方法,规定单个杂质含量小于0.5%,总杂质含量小于1.0%。参考2012版《兽药研究技术指导原则汇编》中“兽用化学药物杂质研究技术指导原则”当杂质有特殊的药理活性时,则杂质的限度可以适当放宽,其制剂的杂质质控限度为1.0%[21]。赵亚萍等[22]制定了ABZ原料药中有关物质检查限度为单杂不超过1.0%,总杂不超过2.0%。刘晓敏等[23]制定了丁二磺酸腺苷蛋氨酸肠溶片的质量标准,其中规定杂质甲硫腺苷的含量不得大于2.0%,其他已知杂质不得大于0.5%,单个未知杂质含量不得大于0.2%,未知杂质总含量不得大于1.0%。因此最终确定本混悬液杂质B(阿苯达唑亚砜)含量不得超过1.0%,其他已知杂质含量不得大于0.5%,且已知杂质总和不得大于2.0%;单个未知杂质含量不得大于0.5%,未知杂质总和不得大于1.0%。
