基于三维卷积神经网络的RNA结构上镁离子结合位点预测
2021-12-17赵彦彭巩卫康刘洋王京京李春华
赵彦彭 巩卫康 刘洋 王京京 李春华
0 引言
自从发现RNA可以催化化学反应以来,它在分子生物学中的重要性不断增加[1],现在RNA被认为在许多细胞调控网络中扮演关键角色[2]。基因表达和调控的实现常常依赖于RNA分子特有的结构和动力学。由于RNA的聚阴离子性质[3],RNA折叠会导致大量负电荷的堆积和链内的强库仑排斥,因此RNA折叠会吸引溶液中的金属离子与其结合,从而降低静电能垒,使折叠后的RNA结构更加稳定。Mg2+在RNA中普遍存在,长期以来被认为是最重要的稳定RNA结构的离子[3],也被认为是RNA能否正确折叠的关键因素。另外,Mg2+的分布也能够极大地影响RNA的局部结构,并导致不同的局部动力学,这一现象和Mg2+的浓度高度相关[4]。例如,TPP-依赖核糖开关功能的发挥不仅受该小分子的调控,还强烈依赖于溶液中Mg2+的浓度[5]。除了稳定结构外,Mg2+还可以帮助核酶执行催化功能[6]。Mg2+对核酶的催化有三重作用:促使核酶折叠成活性三维结构,配合底物和核酶的官能团来定位反应物,水合Mg2+提供或接受质子以促进催化。通过上述作用,Mg2+可提升核酶的催化效率达1011倍[7]。
在过去的十年中,许多高分辨率的RNA结构被解析出来,特别是磁共振(nuclear magnetic resonance,NMR)和单分子荧光共振能量转移光谱技术,提供了RNA折叠和构象变化动力学数据[8]。然而实验确定RNA结构中的离子却存在诸多困难:大多数阳离子在光谱上无法被观察到或者很难被探测到[9];其次由于Mg2+和H2O都有10个电子,所以结合Mg2+很容易被误认为是水分子;再有Mg2+的位置也很难用NMR来确定。因此开发理论预测方法,从RNA序列和结构信息来识别可能的Mg2+结合位点对于理解RNA的结构及功能机制非常重要。
目前预测离子结合位点的工作主要集中在蛋白质上,在RNA上的工作很少。2016年,Hu等[10]提出了基于支持向量机的IonCom方法,用于预测蛋白质上常见的13种金属离子和酸性自由基结合位点。2021年,Snchez-Aparicio等[11]基于金属结合位点在蛋白质骨架结构上出现的偏好性,发展了BioMetAll方法来预测具有特定氨基酸基序的金属结合位点。在RNA方面,2012年,Philips等[12]发展了MetalionRNA方法来预测金属离子(镁、钠和钾)所在位置,该方法基于已知结构中获得的金属离子结合位点的统计势。2018年,Wang等[13]提出RBind方法,使用基于结构的核苷酸相互作用网络方法预测RNA结合位点。RNA的三级结构被转化为一个网络,核苷酸作为节点,它们之间的非共价相互作用作为边。然后,计算了核苷酸的度中心性(degree)和接近中心性(closeness)以确定结合位点。2021年,Su等[14]提出了基于随机森林机器学习方法的RNAsite模型来预测RNA-小分子/离子结合位点,方法是将RNA结构的Laplacian范数、拓扑特性以及溶剂可及性作为特征。然而,在RBind与RNAsite方法中,金属离子结合位点预测的精度低于其他小分子结合位点。这可能是因为许多金属离子往往位于RNA结构的表面,并没有形成形状良好的结合口袋,使其更难预测。另一个可能的原因是训练结构以非金属离子小分子为主,这说明需要发展离子特异性方法来预测金属离子结合位点。
目前深度学习方法由于其具有能够直接从原始数据中提取特征的优势已开始在生物分子结构功能预测领域得到应用。其中,三维卷积神经网络(three-dimensional convolutional neural network,3D-CNN)模型可以将结构转换为多个三维网格数据通道作为输入信息,能够很好处理特征的高维性、不均匀性和方向信息丢失等问题。Li等[15]开发了RNA3DCNN方法,通过将结构三维网格化,提取网格中原子的个数、质量以及电荷信息,来评价RNA三维结构预测的质量。Kozlovskii等[16]开发了BiteNet方法,提取结构三维网格化后的原子密度信息来识别药物结合位点。随着RNA结构数据的增加,基于深度学习预测金属离子结合位点成为可能。
在本研究中,课题组提出了预测RNA三维结构中Mg2+结合位点的方法RNAMg。首先建立了RNA-Mg2+结合位点数据库,随后建立局部坐标系提取局部RNA结构,并用3D图像表示。提取原子类型和电荷量通道信息,通过3D-CNN模型进行学习并预测。通过独立测试集评价了模型的性能,并与最先进的方法进行比较。
1 研究方法
RNAMg以RNA结构中的每个核苷酸为样本预测其是否结合Mg2+。核苷酸及其周围一定范围的微环境被视为3D图像,建立3D-CNN模型进行训练以及预测,模型输出核苷酸是否是Mg2+结合位点。主要工作包括数据集的建立、特征提取、3D-CNN的架构和配置以及评价指标。
1.1 数据处理与分析
首先从蛋白质数据库(protein data bank,PDB)[17]下载所有的共计1 528个只含有RNA分子的结构(2021年1月),之后筛选符合以下条件的结构:(1)结构分辨率优于3.0 Å(10 Å=1 nm);(2)结构中至少含有一个镁离子;(3)对NMR结构,保留PDB文件中的第一个构象;(4)序列一致性小于90%,对于序列一致性大于90%的RNA组,只保留其中分辨率最高的结构[12]。最后,数据库由113个RNA结构组成。
为了能够与目前最先进的方法进行比较,本工作采用了RNAsite中正负样本的定义:如果核苷酸任一原子与Mg2+距离不大于截断距离(4.0 Å),则定义这个核苷酸是正样本,标签设置为1,反之被定义为负样本,标签设置为0。根据以上定义,从数据库所有结构中收集到正样本1 888个,负样本11 504个。由于负样本数量远远多于正样本,课题组使用欠抽样(under-sampling)方法随机减少负样本数量从而实现样本均衡。
使用RNAsite独立测试集中含有Mg2+的结构作为本工作的独立测试集。RNAsite使用以下筛选标准:两个RNA结构之间的TM-scoreRNA[18]大于0.3,则保留阳性样本比例较高的那一个(定义为一个RNA中阳性样本的总数除以序列长度),结果从712条RNA链中筛选得到了78条RNA链。这78个结构以30%的序列相似性作为条件,聚类为57个簇。根据聚类信息对这些RNA进行分割:选取42个包含60个RNA的簇作为训练集,选取15个包含18个RNA的簇作为独立测试集。在独立测试集的18个结构中,有4个结构含有Mg2+结合位点,所以课题组使用这4个结构作为独立测试集,结构信息见表1,它们与本研究的113个RNA的序列一致性均小于90%。它们被用于对模型的测试。
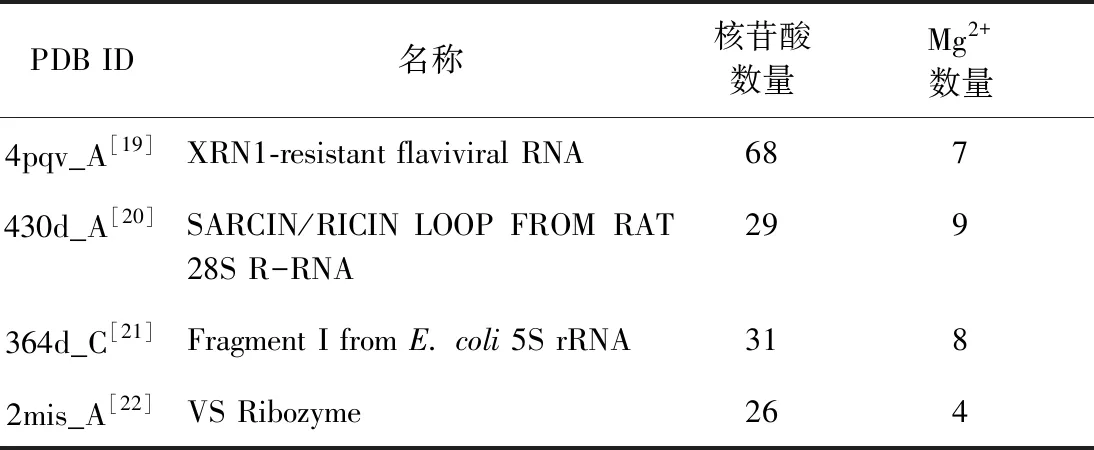
表1 独立测试集中的结构Table 1 Structures in the independent test set
对训练集中的1 888个Mg2+结合位点进行微环境分析,统计了最接近离子的原子类型的分布,图1列出了频数大于2的原子类型分布情况。从图1可以看出,频数最高的原子类型是磷酸盐上的氧原子OP1和OP2,它们是带负电荷的RNA原子,通常需要正电荷来补偿,因此出现频数最高。相比于OP1,OP2更倾向与Mg2+结合。此外,在碱基中更倾向结合离子的原子是O6、N7和O4,特别是O6与OP1的结合频数十分接近。五碳糖中的O3’和O2’也有较强的结合离子的能力。
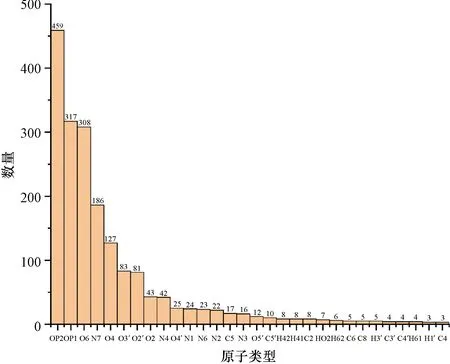
图1 结合核苷酸原子中距离Mg2+最近的原子类型分布Figure 1 Distribution of atom types closest to Mg2+in binding nucleotides
1.2 特征提取
核苷酸周围的环境指的是它的周围的原子分布。课题组建立局部笛卡尔坐标系来确定核苷酸的原子位置。为了获得更好的预测方法,本文构建了两种局部坐标系,分别是以核苷酸的C1’原子和OP2原子作为原点。使用C1’作为原点的原因是该原子的特殊位置:连接了核苷酸的碱基与五碳糖。OP2作为原点的原因是其最偏向与Mg2+结合。另外,尝试了两种不同的特征组合,分别是碳、氧、磷、氮4种原子类型通道模型,和4种原子类型加电荷量的5通道模型。为此共构建4种模型:局部坐标系以OP2为原点并使用4种原子类型通道(OP2+4通道),OP2为原点并使用4种原子类型和电荷量通道(OP2+5通道),以C1’为原点并使用4种原子类型通道(C1’+4通道),C1’为原点并使用4种原子类型和电荷量通道(C1’+5通道)。
坐标系由C1’、O5’、C5’、OP2和嘧啶的N1原子或嘌呤的N9原子的位置确定。具体来说,根据局部坐标系的不同,原点位于C1’或者OP2所在坐标,x轴、y轴和z轴分别用x、y、z表示,满足式(1)~式(3),其中rC1’、rO5’、rC5’和rN表示全局坐标系中从原点指向原子C1’、O5’、C5’以及N1(嘧啶)或者N9(嘌呤)的向量。
x=rN-rC1′
(1)
(2)
(3)
OP2原子只作为局部坐标系原点,其x轴、y轴和z轴的单位向量的计算与OP2无关,只由C1’、O5’、C5’以及N1或者N9原子的位置决定。核苷酸周围的环境包含长度为20 Å的立方体中的原子,该立方体以这个核苷酸的C1’或者OP2原子为中心。这样构建坐标系的目的是保持环境所在的立方体与核苷酸的方向具有一定的一致性,便于特征学习。
随后,每个边长20 Å的盒子被分割成边长1 Å的正方形3D格点,也称作体积像素(简称体素)。对每个体素,用原子类型通道记录碳、氧、磷和氮原子以及电荷量的存在。电荷量从Gromacs软件的amber03力场中提取。对于体素中的一个原子,如N,表示N的通道在其相应位置的值为1,其他原子通道相应位置的值为0,同时电荷量通道对应位置为该原子的电荷量。由此产生的5个3D矩阵作为输入通道堆叠在一起,如图2所示。需要说明的是,对于氢原子,课题组并没有建立相应的通道,因为其位置几乎由其他重原子的位置确定,因此它们在所建通道中已隐式表示。体素选择1 Å可以确保每个体素只能容纳一个原子,从而使网格有更好的空间分辨率。这些通道数据在输入到深度学习网络之前,样本都需要进行零均值归一化。对于每种类型的通道,计算训练数据集中每种类型的通道中所有位置的平均值,并从训练、验证和测试样本中减去。

图2 Mg2+结合位点周围微环境的特征化Figure 2 Featurization of the microenvironment around a Mg2+ binding site
1.3 CNN
图3展示了RNAMg的架构。该神经网络有4个三维卷积层,每个卷积层中的滤波器数量分别为32、64、128以及128,所有卷积层中滤波器的大小为3×3×3体素,卷积步长设置为一个体素。在两个连续的卷积层之后放置一个步长为2个体素的最大池化层。随后,在卷积层之后是一个全连通层,包含512个隐藏单元。最后的输出层是被训练或者预测的样本即核苷酸是金属离子结合位点的概率。隐含层的所有单元均由ReLU非线性函数激活,输出层使用Sigmoid非线性函数激活。

图3 3D-CNN网络结构Figure 3 3D-CNN architecture
训练神经网络的目的是减少真实标签和预测值之间的交叉熵。网络采用Adam算法,通过反向传播对网络参数进行优化。对所有的卷积层和全连通层添加dropout层和正则化层,训练的dropout率为0.5。网络采用截断正态分布初始化权值,使用128的批次大小,运行了200次迭代。
1.4 性能评价
RNAMg模型通过5倍交叉验证(5-fold cross-validation,5CV)训练,并采用独立数据集测试模型的有效性。通过精确率(Precision)、召回率(Recall)和Matthews相关系数(Matthews correlation coefficient,MCC)以及接受者操作特性曲线(receiver operating characteristic curve,ROC)下面积(area under the ROC curve,AUC)来评估分类器的预测性能,其定义如下:
(4)
(5)
MCC=
(6)
通过将每个核苷酸的预测标签与实际标签进行比较,得到真阳性(TP)、假阳性(FP)、真阴性(TN)和假阴性(FN)。其中,Precision是指在所有模型分类为“阳性”的样本中,确实是阳性的占比;Recall是指在所有确实为阳性的样本中,被模型分类为“阳性”的占比;MCC描述的是实际分类与预测分类之间的相关系数,它的取值范围为-1到1,取值为1时表示对测试对象的完美预测,取值为0时表示预测的结果几乎等于随机预测的结果,-1是指预测分类和实际分类完全不一致。课题组还使用接受者操作特性曲线说明模型的性能,计算AUC来评估模型的准确性。AUC分数范围为0到1,AUC =1代表完美分类器,AUC =0.5时是随机分类器。
2 结果与讨论
2.1 特征贡献分析
4种模型在五折交叉验证中的性能如表2所示,其中OP2+5通道模型的Precision、Recall、MCC和AUC结果为0.661、0.668、0.325以及0.724,比C1’+5通道的方法分别提高了4.0%、24.6%、45.1%以及10.7%。这说明选择OP2作为局部坐标系的原点更能突出离子结合位点与非结合位点周围微环境的差别。另一方面,OP2+5通道模型在Precision、MCC和AUC 3个评价指标上都略优于OP2+4通道模型,只有Recall稍稍低于OP2+4通道模型的结果。说明电荷量与原子类型特征在Mg2+结合位点预测方面是互补的,增加电荷量通道有助于RNAMg更准确地预测金属离子结合位点。总体来说,以OP2为局部坐标系原点,使用碳、氧、磷、氮原子类型+电荷量的5通道模型(OP2+5通道)取得了最佳的结果。

表2 考虑不同特征组合与不同局部坐标系原点的3D-CNN模型进行5CV的结果Table 2 Average results of 5CV of 3D-CNN models with different feature combinations and different local coordinate system origins
2.2 模型在独立测试集上的表现
选择OP2+5通道模型作为最终的预测模型,并在独立测试集上测试该模型,结果如表3所示。此外,表3还对比展示了Wang提出的预测工具RBind以及Su提出的预测服务器RNAsite在独立测试集的测试结果。从表3看出本方法在所有评价指标上都略优于另两个方法。RNAMg方法的Precision、Recall、MCC和AUC均高于RBind以及RNAsite方法的相应结果。尤其在MCC上,指标提升明显。综上所述,在Mg2+结合位点预测上,RNAMg模型好于RBind与RNAsite。课题组认为这主要归功于3D-CNN能够直接从初始原子分布中学习更多有用的特征。

表3 RNAMg与其他方法在独立测试集上的比较Table 3 Comparison between RNAMg and other methods on the independent test set
2.3 样例分析
图4显示了在长度为68 nt的4PQV_A上的Mg2+预测结果。该结构有9个Mg2+,7个离子距离RNA结构在4 Å以内,其中的5个Mg2+分别与2个核苷酸结合,2个Mg2+分别与1个核苷酸结合,所以共有12个结合位点。RNAMg成功预测出了12个结合位点中的8个。RNAMg在这个结构上得到的Precision、Recall、MCC和AUC分别为0.363、0.667、0.340和0.749。

黑色表示RNA结构,绿色、红色和蓝色分别表示TP、FP和FN。图4 4PQV_A结构上预测的Mg2+结合位点Figure 4 Mg2+ ion binding site prediction on 4PQV_A structure
3 结论
课题组发展了一种有效预测RNA结构中Mg2+结合位点的理论新方法RNAMg。该方法通过构建基于待预测核苷酸的局部坐标系,从已知RNA结构中提取离子结合位点周围原子分布以及电荷量作为特征,将RNA结构作为3D图像,使用三维卷积神经网络模型进行预测。通过5倍交叉验证测试找到了表现最好的局部坐标系以及特征组合,并证明RNAMg可以准确预测Mg2+结合位点。
目前PDB数据库中具有离子结合的RNA结构数据还非常有限,这阻碍了深度学习预测金属离子结合位点的性能。随着实验解析RNA结构的增多,离子结合位点的数据会不断增长,可以预计深度学习预测模型会不断得到优化,模型的预测能力将不断提升。在独立测试集上的与其他最先进方法的对比证明,RNAMg可以相对准确地预测Mg2+在RNA上的位置,其预测能力优于最先进的预测服务器RBind以及RNAsite,是一种实用的工具。这项工作对理解RNA折叠、核酶催化反应等各种关键生物学过程及相关疾病的产生机制有重要意义。
