RPS6KA3基因变异所致Coffin-Lowry综合征的分子诊断及家系遗传分析
2021-11-06刘渊曾玉坤刘玲黄演林余丽华丁红珂
刘渊 曾玉坤 刘玲 黄演林 余丽华 丁红珂
(广东省妇幼保健院医学遗传中心,广东 广州 511442)
Coffin-Lowry综合征(Coffin-Lowry syndrome,CLS)(OMIM#303600)是一种罕见的X连锁显性遗传病,发病率为1∶100 000~1∶50 000间,以男性患者居多[1]。主要为男性受累,临床症状主要为特殊面容、肌张力减退、进行性痉挛性截瘫、阵发性运动障碍、睡眠呼吸暂停、进行性脊柱侧凸、心血管疾病、骨骼畸形、感音神经性耳聋、视力异常、胼胝体变薄和发育不全、小脑蚓部发育不全、脑室周围白质异常,受影响的男性有较严重的智力障碍,受影响的女性往往有轻度到中度的智力障碍但也可能智力正常,也可能有典型的面部、手部和骨骼症状[1,2]。CLS的致病基因是核糖体蛋白S6激酶多肽3基因(ribosomal protein S 6 kinase polypeptide 3,RPS6KA3),位于Xp22.2,已有报道的RPS6KA3相关变异多达140余种,其中70%~80%为新发变异,家系病例报道相对较少[1]。由于CLS病例大多为新发病例,并无家系关联,且早期的患儿临床表型并无特异,常规的遗传检测并不能对该病进行高效诊断。本研究中,我们利用高通量测序技术对2例Coffin-Lowry综合征家系进行变异基因检测及分析,并对检测到的变异位点进行Sanger测序验证,对Coffin-Lowry综合征患者进行了分子诊断,丰富了Coffin-Lowry综合征的病因信息,同时为该家庭的临床诊断和遗传咨询提供了遗传学证据。
1 材料与方法
1.1 病历资料
1.1.1 先证者1 男性,现1岁,足月顺产,出生体重3.0kg,出生时无窒息抢救史。胎儿期发育持续小于孕周2~3周。出生3个月后听力筛查未通过,且抬头不稳。体格检查发现患儿特殊面容(眼距宽、鼻梁扁平、牙间距大);仅能扶物站立,尚无语言发育,精神运动发育迟缓。
家系调查:先证者母亲未见明显异常,因再次怀孕,来本中心就诊。经遗传咨询后,家属知情选择对先证者及父母进行医学外显子检测,经高通量测序及Sanger测序验证先证者致病基因为RPS6KA3新发半合子变异[c.1672C>T(p.R558*)]。孕妇知情选择行羊水穿刺,利用羊水DNA对当前胎儿检测RPS6KA3基因c.1672位点是否与先证者相同,同时行羊水染色体微阵列分析(chromosomal microarray analysis,CMA)及荧光定量聚合酶链反应(quantitative fluorescence polymerase chain reaction,QF-PCR)检测以排除胎儿染色体异常。
1.1.2 先证者2 男性,现4岁,患儿出生时哭声弱、听力筛查未通过;家属告知4个月大时患儿表现为竖头不稳、不能翻身、肢体活动偏少、肌张力低下、自主意识欠佳;患儿表现为精神运动发育迟缓、特殊面容、眼距宽、鼻梁平、极重度耳聋。曾行头部磁共振成像(magnetic resonance imaging,MRI)检查,提示脑室增宽、脑外间隙增宽、脑萎缩等。高通量测序结果显示患儿为RPS6KA3新发半合子变异(c.325+2_325+3insT),现要求针对先证者RPS6KA3基因变异位点进行产前诊断。
家系调查:先证者母亲现30岁,孕16周,未见明显异常。经遗传咨询后,家属知情选择对先证者RPS6KA3基因变异位点进行家系验证。孕妇知情选择行羊水穿刺,利用羊水DNA对当前胎儿检测RPS6KA3基因的先证者变异位点,同时行羊水CMA及QF-PCR检测以排除胎儿染色体异常。
本文所涉及研究获得了广东省妇幼保健院机构审查委员会及伦理委员会的批准。所有样品的采集均取得了参与者的书面知情同意书。
1.2 实验方法
1.2.1 外周血及羊水DNA提取 经知情同意后,采集先证者及其父母外周血2ml(EDTA抗凝);孕妇在超声引导下进行羊水穿刺,抽取20ml羊水,所采集样本及时送到实验室处理。利用柱提法对外周血和羊水进行基因组DNA提取,所用试剂盒为德国Qiagen公司生产的Qiamp DNA Blood Mini Kit。提取完成后,用NanoDrop2000超微量分光光度计及Qubit定量试剂盒(Thermo Fisher)对DNA进行纯度和浓度的测定,合格DNA保存于-20℃备用。
1.2.2 羊水染色体微阵列分析 通过快速QFPCR法对D13S305、D18S978、D21S11等11个STR位点,分析胎儿染色体非整倍体情况,及排除采集胎儿标本无母体DNA污染。采用美国Affymetrix公司的单核苷酸多态性微阵列检测平台(Affymetrix CYTOSCAN 750K),取300 ng基因组DNA按照实验标准流程进行酶切、连接、PCR、纯化、片段化、标记、杂交、洗涤、扫描。运用4.2版的Chromosome Analysis Suite软件进行分析。结果判读参考人类孟德尔遗传在线数据库(OnlineMendelian Inheritance in Man,OMIM)、DECIPHER数据库、临床基因组变异数据库(Clin Var)、Database of Genomic Variants数据库和在临床基因组资源(clinical genome resource,ClinGen)等数据库综合分析。
1.2.3 二代测序 基因组DNA经片段化、连接接头、扩增纯化后进行液相探针捕获(illuminaTruSeq protocol),捕获探针覆盖了具有已知功能的4000种临床致病基因,其中包含50 584个编码区和8 421 879个碱基对。平均测序深度为300X以上,最高测序深度为500X;大于10X的覆盖范围占致病基因的98.9%,大于20X的覆盖范围占致病基因的98.5%。使用HiSeq2000测序仪(Illumina,Inc.,San Diego,CA)进行测序。所得原始数据经过质检、过滤、序列比对及变异注释等生物信息学分析。
1.2.4 Sanger测序 根据高通量测序结果,采用Primer 5软件设计引物,用先证者及父母外周血DNA进行变异位点Sanger测序验证,并进一步对孕妇胎儿是否携带相同位点进行产前诊断。一代测序的结果与数据库的序列进行比对,确定二代测序发现的疑似致病变异。参考序列数据来自数据库UCSC Genome Browser(http:∥genome.ucsc.edu),序列版本号:GRCh37/hg 19。
家系1变异,上游引物:AGGTCAGCACTCAT CATCTTG,下游引物:TGAAAGGCTGGCATTT TATC;
家系2变异,上游引物:TGTATTATAGCAG GGTCTTATTTTAAC,下游引物:ATAACATGC CAAGGAGTCCC
2 结果
2.1 羊水染色体微阵列分析及QF-PCR结果 提示胎儿染色体未出现非整倍体及微缺失和微重复异常。
2.2 二代测序及家系验证 家系1中,父母子核心家系医学外显测序发现先证者RPS6KA3基因发生变异c.1672C>T(p.R558*),先证者父母未发现该变异位点(图1)。家系2中,父母子核心家系医学外显测序发现先证者RPS6KA3基因发生变异c.325+2_325+3insT,先证者父母未发现该变异位点。

图1 先证者1家系的高通量测序结果(RPS6KA3基因为反义链编码基因)
2.3 一代测序验证及产前诊断结果 Sanger测序验证结果显示,先证者1的RPS6KA3基因发生变异,c.1672C>T(p.R558*)半合子,为无义变异(图2)。
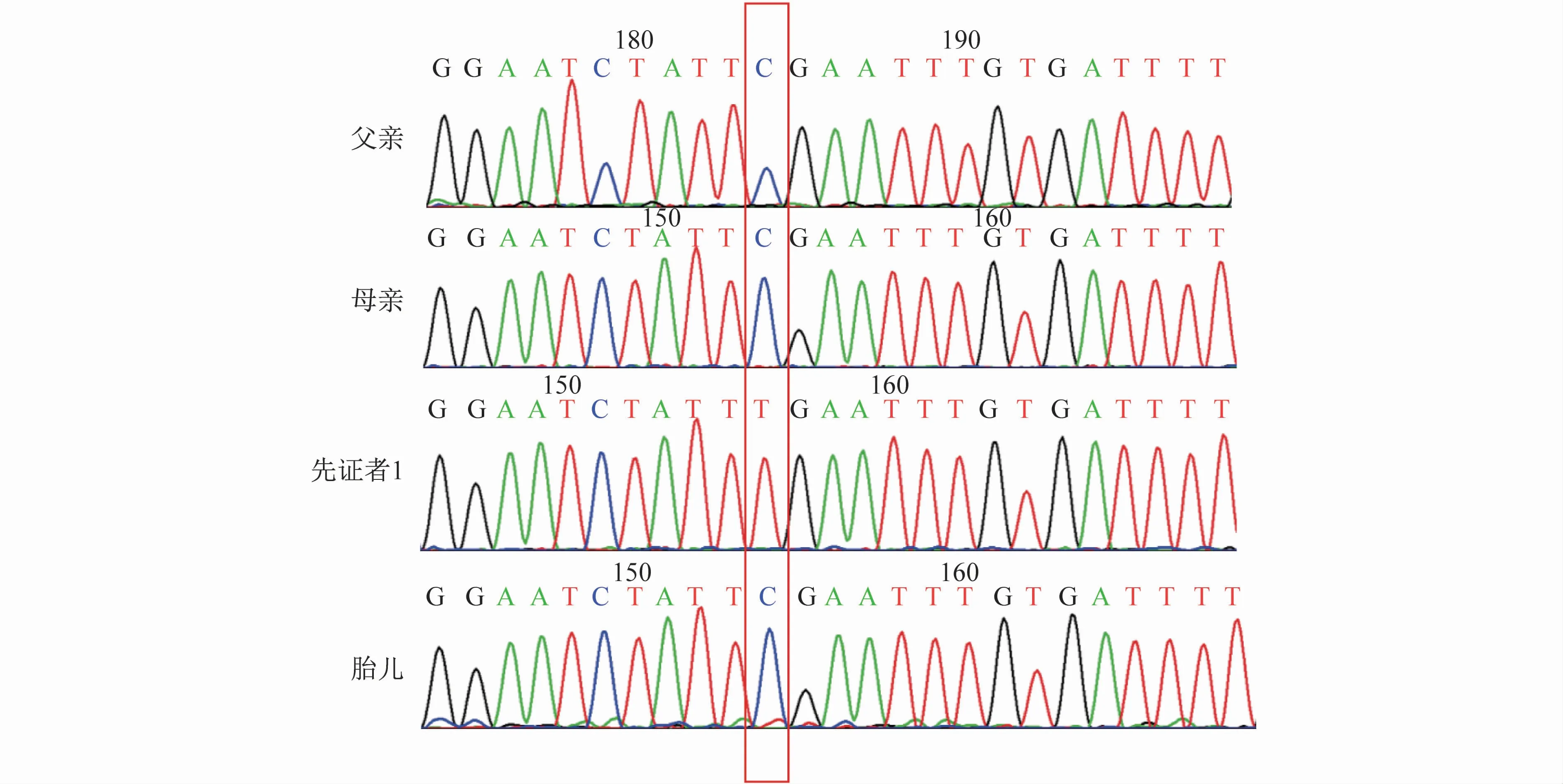
图2 家系1父母子及胎儿一代测序结果(上游引物:AGGTCAGCACTCATCATCTTG,下游引物:TGAAAGGCTGGCATTTTATC)
先证者2在c.325+2_325+3insT发生半合子移码变异(图3),先证者父亲、母亲及胎儿该位点均未发现先证者变异类型,两个家系中的先证者判断均为新发变异。对当前胎儿均进行了CMA检测,结果正常,经遗传咨询,先证者父母决定保留胎儿,截至该文撰稿时未发现异常情况。
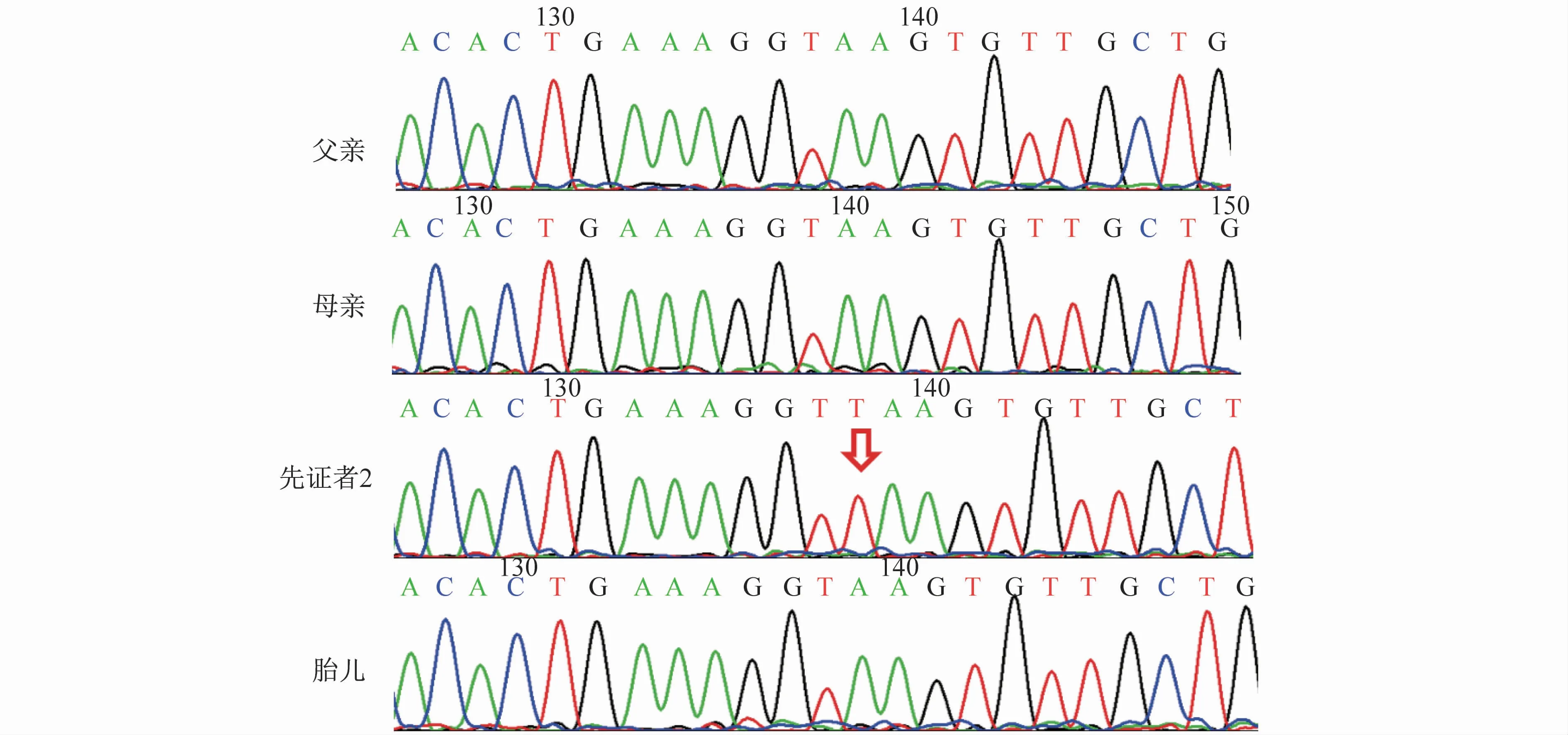
图3 家系2父母子及胎儿一代测序结果(上游引物:TGTATTATAGCAGGGTCTTATTTTAAC,下游引物:ATAACATGCCAAGGAGTCCC)
2.4 先证者1岁前后表型对比见表1。

表1 先证者1岁前后出现的表型
3 讨论
对CLS病例的首次描述是Coffin等[3]在1966年完成的。此后,对该综合征的基础研究证实是由于RPS6KA3基因变异所致[1]。RPS6KA3位于X染色体短臂,含有22个外显子,编码产物为丝氨酸/苏氨酸激酶rsk(核糖体s6激酶)家族的一个成员,该激酶包含740个氨基酸(90 kDa),共有2个不同的激酶催化结构域,该激酶是RAS-RAF-MEK-ERK信号通路中MAP激酶ERK下游重要效应分子之一,广泛表达于哺乳动物并控制细胞的生长和分化,已有研究在小鼠和果蝇模型中证实了该激酶对神经元功能的影响,这可能与CLS患儿的精神运动功能障碍有关[4,5]。RPS6KA3具有很强的等位基因异质性,迄今已在CLS患者中鉴定出140多个不同的失活变异,其中大约30%的变异是错义变异,15%的为无义变异,20%的是剪接错误和30%的微小缺失或插入[1]。
目前,CLS确诊主要依赖于患儿临床表现和分子诊断,临床表现包括面部畸形(如前额突起、眼距增宽、鼻梁扁平、厚唇外翻等)、精神运动发育迟缓、语言听力障碍、骨骼畸形等[1,6]。对于CLS患者来说,越早诊断对于患者的正确治疗越有帮助,包括监测某些特定并发症和提高患儿生活质量等方面都至关重要[1]。虽然表型对CLS的提示有很大帮助,但对临床医生的经验要求较高,即使高度怀疑也无法做出精准诊断。另一方面,在CLS患儿出生后的首年内,大多生长发育参数在正常范围,其他体征并不明显,也不特异,常仅伴有关节张力低下和关节松弛表现,并不会被注意到,这些都提高了对CLS患儿早期诊断的难度[7]。本文所描述2例先证者均在1岁后才出现典型的面部特征和精神运动发育异常(见表1)。
随着分子技术的发展,遗传病的诊断越来越多地采用更先进的分子遗传学检测技术,可对患者进行早期诊断和监测。从最近几年的CLS病例来看,多依赖高通量测序技术诊断[8-15],也有用其他分子方法对覆盖RPS6KA3基因区域的染色体微小重复的诊断[16,17]。
本报道描述的第一个家系中,先证者转诊到我中心时已有1岁,问诊发现先证者在胎儿期即有表现出生长发育小于孕周2~3周,但当时未行进一步的遗传病诊断,足月出生时也无特殊异常。患儿出生3个月后也表现出相关发育迟缓,未找到病因,患儿母亲又再次怀孕,患者父母心理负担极大。到本中心就诊时,患儿已表现特殊面容明显(眼距宽、鼻梁扁平、牙间距大),语言精神运动发育明显迟缓,提示遗传病可能性大。高通量测序及Sanger测序验证先证者致病基因为RPS6KA3半合子变异,确定患儿为CLS患者,且为新发变异[c.1672C>T(p.R558*)](图2)。对于二代测序结果再次咨询后,孕妇选择了羊水穿刺手术,利用羊水DNA对当前胎儿RPS6KA3基因进行了检测,并未发现与先证者相同变异。同时,为排除胎儿染色体数目异常及染色体微缺失和微重复综合征,对当前胎儿同时进行了快速QF-PCR检测及羊水染色体微阵列分析,结果均为正常。本病例中变异位点RPS6KA3[c.1672C>T(p.R558*)]为无义变异,早前国外有过1例相同位点的报道[18],本研究为中国人群首次报道此变异位点。
第二个家系中,先证者为RPS6KA31新发半合子变异(c.325+2_325+3insT)(图3),此位点的变异也有过报道[19],本文为中国人群也是首次报道,家系验证显示患儿为新发变异。据患儿父母提供资料显示,该患儿出生时哭声弱、听力筛查未通过;4个月时显示肌张力低下;患儿现4岁,主要表现为精神运动发育迟缓、特殊面容、眼距宽、鼻梁平、极重度耳聋。患儿曾多次行头部MRI检查,提示脑室增宽、脑外间隙增宽、脑萎缩等,为CLS常见表型。患儿母亲因再次怀孕前来咨询,家系验证显示先证者为新发变异,同时父母选择对本次妊娠行羊水穿刺术,利用羊水DNA对当前胎儿RPS6KA3基因验证,结果显示胎儿RPS6KA3基因未检测到c.325+2_325+3insT变异,未重复先证者基因型。对当前胎儿同时进行了快速QF-PCR检测及羊水染色体微阵列分析,结果也均为正常。
对于发现CLS患者家庭,先证者RPS6KA3变异来源决定了遗传咨询和指导的决策,对家庭再生育的指导至关重要。如患儿的变异来自携带者母亲,该母亲即存在50%风险将该变异遗传给下一代,下一代如果是男性,则为CLS患者,如为女性,则会出现CLS相关发育异常高风险[1]。本研究中,通过二代测序技术,我们发现了2例CLS病例,通过家系验证并未发现患儿父母携带先证者变异,判断2例患儿均为新发变异,夫妻目前再次生育CLS患儿风险低。考虑到母亲存在生殖腺嵌合的可能,对患儿母亲再次怀孕也进行了RPS6KA3基因产前诊断,均未检测到与先证者相同变异,同时也为胎儿做出了常规的染色体微缺失微重复和常见染色体非整倍体检测,为此2个家庭做出了关键的生育指导。近年来,二代测序技术(next generation sequencing,NGS)在罕见遗传病领域的诊断在学术界已基本达成共识,在国内外得到广泛应用。如果患者的表型特异性强,符合某一类遗传病,可使用基因靶向测序,如耳聋基因靶向测序;但很多遗传病,如CLS,早期表型(1岁以前)不明显,且常规的检测无法找到病因,可考虑用全外显子测序或全基因组测序。
