卤醇脱卤酶的研究进展
2021-11-05王龙兴贾红华
王龙兴,贾红华,韦 萍
(南京工业大学 生物与制药工程学院,江苏 南京 211800)

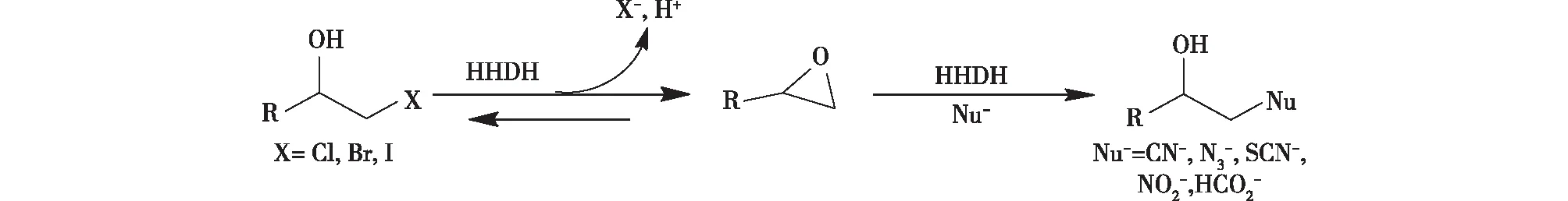
图1 HHDHs催化的脱卤与开环反应
在早期的研究中,利用产HHDH的微生物来降解环境中含有卤代有机化合物的污染物,使其在环境治理方面具有重要应用价值。之后,通过对HheC突变、结构解析以及动力学等方面的研究,揭示了HHDH的结构特点以及催化脱卤、开环的机制[8-9],发现HHDHs在催化制备手性环氧化物和光学纯的β-取代醇方面具有极高的应用价值[10],尤其是在制备降脂药阿托他汀的前体、手性环氧氯丙烷等方面,这使得HHDHs应用前景更广阔。因此,本文综述了近年来关于HHDHs在来源分布、结构与机制、分子改造以及生物催化应用等方面的研究进展,为相关的研究者提供参考。
1 HHDHs的来源与分布
HHDHs从发现到酶家族的扩展,经过了几十年的发展,最初仅发现了HheA、HheA2、HheB、HheB2和HheC这5种代表性的HHDHs。它们分别来自Corynebacteriumsp. N-1074[11]、Arthrobactersp. AD2[8]、Arthrobactersp. AD2[11]、Arthrobactersp. AD[8]、AgrobacteriumradiobacterAD1[8]和Rhizobiumsp. NHG3[12]。这5种酶又被分A、B和C三个亚类,亚类内的酶序列相似度超过97%,亚类间的相似性却低于33%,但与SDR家族其他酶具有较高的相似性。
近年来,随着各类数据库中基因序列的爆发式增长,通过生物信息学手段挖掘新酶已成为主要途径[13],因而HHDHs酶家族的数量迅速扩增。目前文献中已报道的HHDHs达到77种之多[14-17],其中已有20多种酶得到详细的表征,不同酶的底物谱及立体选择性都有差异(表1),大多数HHDHs脱溴的活力高于脱氯的活力,这是由于C—Cl的键能高于C—Br,使得C—Cl不易断裂;另外,有研究表明HHDHs不能催化C—F断裂而脱氟[18]。

表1 已表征HHDHs的底物谱及活性
根据进化分析,HHDHs酶家族已由A类扩增到G类,共7个亚类,各亚类之间的HHDHs相似性为25%~45%。HHDHs家族的扩增主要集中在A、B、D和E亚类,虽然目前HheC是目前研究最多的亚类,但迄今仍然仅一种(图2)。目前发现的HHDHs中,来源于海洋宏基因组的HHDHs集中分布于B类和E类,而其他亚类的HHDHs则多来源于土壤、海洋、污水或淡水淤泥等环境的细菌,其中以变形菌门居多。从HHDHs的来源来看,多为卤素富集区域,这恰与微生物能够在含有有毒的卤代有机物中利用HHDHs代谢卤代醇生长繁殖的习性相适应,暂时还未在其他生物中发现HHDHs的踪迹。

图2 HHDH酶家族进化分析
Koopmeiners等[19]在对17种新酶进行表征的时候发现,酶的立体选择性似乎和酶的分类存在着关联,在对2-氯-苯乙醇进行脱卤反应时,B类、E类新酶的立体选择性为S型,而A类和D类的新酶却是R型。但是Xue等[14-15]对挖掘的新酶进行表征的时候发现,即使同亚类的酶针对同种底物也有不同的立体选择性。就立体选择性而言,HheC仍是催化脱卤反应的对映选择性最高(E>100)的酶[15, 19]。但HheC的热稳定和催化反应的最适温度明显低于HheA3、HheA5、HheD、HheD3和HheD5[19],这也是HheC进行分子改造时的重点研究领域。从新酶的表征数据来看,还未发现HHDHs的分类和功能之间的规律。
2 HHDHs的结构与催化机制
HHDHs的活性中心是由Ser-Tyr-Arg构成的催化三联体,这个结构与SDR家族非常相似,不同的是SDR家族是Ser-Tyr-Lys[7,9]。通过对已报道的HHDHs的氨基酸序列进行多序列比对发现,Tyr与Arg之间总被3个氨基酸隔开[13],而SDR家族中,超过86%酶的Tyr与Lys之间也相隔3个氨基酸[6-7];在HHDHs中,Ser与Tyr之间相隔12个氨基酸,但SDR家族中Ser与Tyr的相对位置却不是那么保守[13]。因而由催化三联体结构组成的S-X12-Y-X3-R基序被认为是辨别HHDHs与SDR家族的重要特征之一(图3)。此外,HHDHs在N端还拥有1个含芳香族氨基酸(Phe或Tyr)的宽敞的阴离子结合口袋,这一结构替代了SDR中富含Ala或Gly的NAD(P)辅酶结合位点的位置,成为另一个辨别HHDHs的特征序列T-X4-(F/Y)-X-G[9](图3)。阴离子结合口袋通过主链上的酰胺键形成的氢键、芳香族氨基酸残基带的正电荷以及水分子的结合来稳定阴离子。虽然在酶的结构中有许多氢键供体,但是芳香族的氨基酸残基能使碳卤键更轻易地断裂[23]。

图3 HHDHs的特征基序
迄今为止,人们已测定了HheA[24]、HheA2[25]、HheB[24]、HheC[9]和HheG[26]的晶体结构,由此发现,HHDHs是由一对二聚体组成的一个同源四聚体(图4)。与SDR家族一样,HHDHs拥有一个典型的罗斯曼折叠结构——7个或者8个α-螺旋包裹着6个或7个平行的β-折叠。虽然酶活性中心在酶的内部,但是底物能通过一个通道与活性中心的氨基酸残基结合,通道中的氨基酸残基则影响HHDHs的底物特异性和产物立体选择性[27-29]。
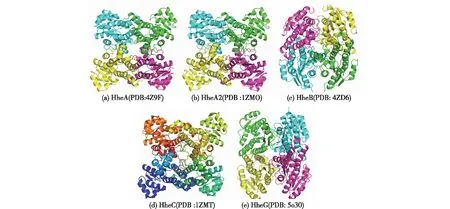
图4 HHDHs的晶体结构


图5 HHDHs的催化机制
3 HHDHs的分子改造
野生型酶的催化性能往往无法适应工业化应用的需求,随着定向进化等技术的发展,通过对HHDHs进行酶分子改造以提升其性能是一种重要途径。HheC在制备阿托他汀药物前体方面具有很高的价值,但野生型酶的立体选择性及酶活力较差。2007年,Fox等[31]首次采用具有革命性的技术——ProSAR驱动的定向进化技术,针对HheC催化(S)-4-氯-3羟基丁酸乙酯转化为(R)-4-氰基-3-羟基丁酸乙酯的反应进行分子改造,在众多的突变体中至少筛选到含有35个突变体,产物的体积生产率提高了4 000倍左右。在该研究的基础上,Schallmey等[29]构建了含有37个突变位点的经典突变酶HheC-2360,通过酶学性质表征和结构解析研究发现,HheC-2360催化(S)-氯-3羟基丁酸乙酯脱卤的kcat提高了3.1倍,在CN-介导下催化(S)-3,4-环氧丁酸酯开环的kcat提高了10倍,立体选择性由HheC的R型转变为HheC-2360的S型,并且热稳定性提高了8 ℃。以HheC-2360的晶体结构为模型进行分子动力学模拟,与产物(3R,5R)-6-氰基-3,5-二羟基己酸叔丁酯进行分子对接,辨识出关键氨基酸残基位点,然后对关键位点进行定点突变,筛选出突变体V84G/W86F,其催化活性比HheC-2360提高了15倍,Km值降低了4/5,kcat增加了3.3倍,使酶与底物的亲和性大大提高并扩大了酶与底物的结合空间,产率提高了2倍[32]。此外,HheAAM(HheA12)通过迭代饱和突变后,突变体M4-HheAAM对(3R,5R)-6-氯-3,5-二羟基己酸叔丁酯的比酶活达到了0.89 U/mg,比野生型提高13倍[33]。
另外,提高HheC的稳定性也是近年来研究的重要内容。汤丽霞团队的Wu等[34]采用组合定向进化的策略,先对野生型HheC进行易错PCR的随机突变,筛选出6株阳性突变体,共包含8个突变点,即F12P、D182E、Q87R、N157V、K52E、M252L、F136I和N159R;然后通过迭代饱和突变对这8个突变位点进行突变,发现随着突变点增加,酶的热稳定性随之提高,最终筛选拥有8个突变位点的突变体ISM-4,在未影响其酶活力的前提下,较野生型的酶,其热稳定性大大提高,65 ℃的半衰期提高了3 400倍,tm提高了18 ℃。依据在HheC的表面选取两个相邻的带电荷且与活性中心距离不超过1.2 nm的原则,Wang等[35]选取K203和K204两个位点同时突变为带有电荷的R、D、E和K,从48个克隆筛选出CSL1 (K203R)、CSL2(K204R)、CSL3 (K203R/K204R)3个阳性突变体;与野生型相比,CSL2在55 ℃的半衰期提高了近6.9倍且不影响酶活性,CSL3在55 ℃的半衰期提高了3.4倍,但是其催化1,3-二氯-2-丙醇的kcat值提高了1.8倍;据动力学模拟分析,这是由于突变点的氨基酸残基变化优化了蛋白质表面电荷间的相互作用和氢键作用,使整体结构更稳定,从而提高HheC的热稳定性。与传统的易错PCR等定向进化技术相比,Arabnejad等[36]采用FRESCO方法,通过能量计算、二硫键预测以及分子动力学模拟对HheC的突变位点进行理性设计,不仅使突变后的HheC热稳定性提高了23 ℃,而且耐有机溶剂;突变体HheC-H12在其最适温度50 ℃下,其活性是野生型的2倍,在50%甲醇、50%乙腈、25%DMSO、25%二恶烷、25%四氢呋喃和25%二甲基甲酰胺溶液中能维持活性不变,其中在50%乙腈溶液中能保持5 h以上无活性损失,且动力学常数和底物范围与野生型相近;HheC-H12包含了12个突变点:A29L、D39K、E64R、S68R、Q87R、A93T、C153N、A158V、E190T、E197K、V99I和V236I,其中C153N对稳定性的贡献最大,它不仅改善了残基间的相互作用力,还在溶剂间形成稳定的氢键,它也成为迄今是在稳定性方面表现最佳的突变体。
HheG是近年来通过基因挖掘发现的新酶,对氧化柠檬烯和环氧环己具有很高的活性,但是其稳定性不佳,限制了它的应用。Solarczek等[37]计算模拟分析HheG的晶体结构,筛选了20个氨基酸残基位点进行饱和突变,最终发现T123位点的突变体T123G、T123F、T123Y、T123W和T123H的tm不仅提高了7~14 ℃,其酶活力还提高了3倍,其中T123G催化反应的初始速度更是提高了5倍。根据分子动力学模拟发现,靠近HheG活性位点裂缝位置有一个快速移动的环,处于将裂缝部分覆盖或不覆盖的状态。当裂缝部分被覆盖的时候,酶活性位点不易于底物接触,而环路迁移率和位置直接受123位氨基酸残基的影响[35]。
4 HHDHs的应用
4.1 HHDH制备阿托他汀药物前体
HHDHs的一个重要应用是催化合成降血脂药阿托他汀侧链基团(R)-4-氰基-3-羟基丁酸乙酯。早期研究采用HheC及其突变体[31]和(S)-型酮还原酶联合催化下合成了(R)-4-氰基-3-羟基丁酸乙酯。而采用HheA的全细胞催化 (S)-4-氯-3-羟基丁酸乙酯转化为(R)-4-氰基-3-羟基丁酸乙酯的转化率和产率分别可达95%和85%,但是细胞的消耗较大[38]。后来采用1,3-二氯-2-丙醇为原料,HheC先催化脱卤并在CN-介导下开环合成(S)-4-氯-3-羟基丁腈,然后再次脱卤开环合成(R)-4-氰基-3-羟基丁酸乙酯[39]。在填充柱中填充HHDH固定化酶,连续催化(S)-4-氯-3-羟基丁酸乙酯合成(R)-4-氰基-3-羟基丁酸乙酯,10 d后底物转化率达90.6%,经过精馏提纯可将收率达到98.2%、纯度达到99.3%、光学纯度达到99.1%[40]。另外,HheC突变体和腈水解酶AtNIT2共同级联催化,可直接将(S)-4-氯-3-羟基丁酸乙酯转化成(R)-3-羟基戊二酸,更有利于后续阿托伐他汀侧链基团的合成[41]。因为(R)-4-氯-3-羟基丁酸乙酯作为中间体合成阿托伐他汀的路径需要-78 ℃的极端条件,Luo等[32]便改进了路径,利用HheC2360-V84G/W86F催化(3S,5R)-6-氯-3,5-二羟基已酸叔丁酯转化为(3S,5R)-6-氰基-3,5-二羟基己酸叔丁酯,从而避免了原先路线后续的苛刻反应条件。
4.2 HHDH制备环氧氯丙烷

4.3 HHDH在生物催化其他方面的应用

5 总结和展望
HHDHs因其既能催化邻卤代醇脱卤成环,又能催化环氧化物开环的特殊机制,使其成为生物催化领域一种重要的生物催化剂。它不仅可用于降解卤代有机污染物,还可广泛应用于降脂药阿托伐他汀前体、环氧氯丙烷以及其他环氧化物、β-取代醇等化合物的制备。它的催化三联体构成的基序S-X12-Y-X3-R以及N端的阴离子口袋构成的基序T-X4-(F/Y)-X-G是辨识HHDH序列的两个重要特征,也是基于数据库挖掘新酶筛选的标志。随着基因测序技术的发展,生物信息数据库中基因数据呈现爆炸式的增长,通过HHDHs序列特征在数据库中挖掘新的HHDHs是当前的主要方向。可以预计未来会有更多的HHDHs家族其他成员被挖掘出来,不断拓展HHDHs的来源。
从目前已表征的酶来看,野生型酶的稳定性和立体选择性不佳,很难满足工业生产的需求。通过ProSAR驱动的定向进化技术、易错PCR、迭代饱和突变、FRESCO法等方法对酶分子改造,HHDH的稳定性和立体选择性均有显著提高。随着蛋白质工程技术的快速发展,定向进化技术越来越成熟以及相应的高通量筛选设备来越先进,预计未来将改造出更多具有优异性能的HHDHs,以满足工业过程的需要。另外,酶固定化技术也提升了HHDHs的催化性能,使HHDH可以回收,多次重复利用,在生物反应器中,可连续性操控生物催化反应,更加适应工业化大规模生产的需求。
由于脱卤反应进行高通量筛选HHDHs的显色试剂以及开环反应所需的亲核试剂多为剧毒、易制爆药品,普通实验室硬件设施难以满足条件,一定程度上阻碍了HHDHs的快速发展。开发安全便捷的分析方法也将是研究HHDHs的重要内容。
随着酶工程技术越来越成熟以及高通量筛选等设备越来越先进,基于数据库挖掘新酶,通过分子改造及固定化等技术获取更多能够满足工业需求的HHDHs,完善HHDH生物催化的工艺条件将成为研究人员未来关注的焦点,预计未来将有许多可适应医药化工等行业大规模生产需求的HHDH商业酶产品。
