从单酶催化到多酶级联催化
——从王义翘教授在酶技术领域的贡献说开去
2021-09-24吴淑可周颐王文张巍高鹏飞李智
吴淑可,周颐,王文,张巍,高鹏飞,李智
(1华中农业大学生命科学技术学院,湖北 武汉 430070;2雀巢新加坡研发中心,新加坡 619625;3新加坡科技研究局(A*STAR)生物过程技术研究所,新加坡 138668;4葛兰素史克中国投资有限公司,北京 100025;5新加坡国立大学化学与分子工程系,新加坡 117585)
麻省理工学院王义翘教授是国际生物工程和生物技术领域的奠基人和业界泰斗,开拓了现代生物化工和生物制药领域新的疆界并引领了其发展。他的研究方向涵盖了生化工程的各个子领域,包括:上游的基因与分子工程(例如酶技术和酶工程)、中游的生化过程工程与工艺(例如高密度培养动物细胞、生产重组抗体药物、计算机控制生化反应过程)、下游的生物分离纯化技术(例如生物膜和色谱技术、糖基化蛋白的质量控制、蛋白质的纯化和复性),以及贯穿上中下游的整合生物过程工程(例如微生物降解纤维素生产生物能源、微生物脱硫技术)等[1]。作为王义翘教授在酶技术领域的学生和合作者,我们非常荣幸能在此介绍王教授早期在酶技术领域的贡献,简述我们在王教授的合作、指导和帮助下获得的酶技术领域的成果,以及延伸讨论多酶级联催化领域的最新进展,并对进一步结合合成生物学的理念和思想[2]的可行性进行展望。
1 王义翘教授早期在酶技术领域的贡献
酶作为生物体内具有催化功能的元件,在生命的诞生、生长、延续、繁衍中起着不可替代的重要作用。从应用角度来讲,酶已经滋养了人类饮食文化上千年,例如微生物酿酒、发酵食品等。伴随着近代以来科技的迅猛发展,酶在社会生活中有着更为广泛的应用,例如食品、能源、化工、制药业、轻工业、农业领域等[3-7]。王教授早期的独立学术生涯始于MIT 的营养及食品科学系。早在20 世纪70 年代,他便利用蛋白酶(枯草芽孢杆菌来源)来消化鱼蛋白固体浓缩物(fish protein concentrate),并研究了该非均相酶促反应的动力学[8],以及对汞含量的影响[9]。基于当时美国社会对利用化石燃料来源的碳一原料的兴趣,王教授研究了源自多形汉逊酵母(Hansenula polymorpha)的甲醇氧化酶,优化了酶提取的过程,开发了以二乙氨基乙基纤维素(DEAEcellulose)为载体的酶固定化方法,并且研究了该酶在分批反应器和连续性反应器中氧化甲醇生产甲醛的过程[10]。20 世纪70—80 年代也是抗生素发掘的黄金年代,当时王教授就提出了利用纯化的多个酶合成天然产物的方案,利用短芽孢杆菌(Bacillus brevisATCC 9999)中部分纯化的非核糖体多肽合成酶(2 个组分),首次实现了无细胞多酶反应制备级全合成约500 mg 的短杆菌肽S(gramicidin S)[11],并且通过与Robert Langer 教授和George Whitesides教授等的合作,实现了该反应中重要的辅因子ATP 的再生[12]。针对这个反应,王教授还开发了短芽孢杆菌的高密度发酵工艺,用于生产非核糖体多肽合成酶[13],并深入研究了该非核糖体多肽合成酶催化的反应动力学[14]。在80 年代以后,王教授的研究重心逐渐转向了动物细胞培养和蛋白药物生产的方向,但仍继续开展一些酶技术相关的科研工作,例如和当时还是博士生的Andreas Bommarius 教授合作研究了黄嘌呤氧化酶(xanthine oxidase)在反胶束体系(reversed micellar systems)中的反应动力学,为氧化还原酶在非水相溶剂中的反应提供了理论基础[15],并利用萤火虫来源的荧光素酶(luciferase)开发了发酵过程中在线检测活细胞数量和胞内ATP浓度的方法[16-17]。通过回顾王教授早期在酶技术领域的贡献,我们可以发现虽然王教授没有同Frances Arnold 教授[18]或者Manfred Reetz 教授[19]那样围绕酶技术的一个重要方向(定向进化)进行全面深入的研究,他仍然在酶技术领域的几个子方向上做出了重要贡献。即使从几十年后的今天来看,他所探寻的诸多方向仍是酶技术的研究热点,例如食品的酶消化[20]、碳一原料的酶转化[21-23]、无细胞多酶生物合成[24]等。更难能可贵的是,王教授在这些不同的酶技术项目中,都非常注重定量的工程化思想,对多种较为复杂的酶催化反应进行了催化动力学和数学建模,这对于今天的酶技术领域的研究和发展都极具借鉴价值和指导意义。
2 王义翘教授在新加坡-麻省理工联盟项目的工作
从20 世纪90 年代起,王教授长期推动并制定了新加坡生物技术和生物医药领域的长期发展规划,担任新加坡国立大学/MIT 淡马锡教授多年,主持创建了新加坡 A*STAR Bioprocessing Technology Institute,并获新加坡李光耀总理颁发的国家公共服务奖章。从2005 年到2015 年,年逾古稀的王教授通过新加坡-麻省理工联盟(Singapore-MIT Alliance)的化学工程与制药工程项 目(Chemical and Pharmaceutical Engineering)与新加坡国立大学的李智教授(本文作者)合作,联合指导了几名博士生(本文作者王文、张巍、高鹏飞、吴淑可),在酶技术领域的几个子方向上继续开展了重要的工作。尤其令我们感动的是,在联合指导的过程中,近80 岁高龄的王教授仍然不辞辛苦,多次从波士顿飞往亚太地区,亲自莅临一线指导科研工作[图1(a)],并积极参与亚太地区的学术会议。在2013 年,他还作为大会荣誉主席出席了北京第18 届国际生物化学和分子工程大会[图1(b)]。联合培养的博士生也有机会来到世界顶尖的MIT化工系,参与到王教授MIT实验室的最后几项研究中[25]。这不仅让我们能亲自了解和领略到化工领域的最前沿进展,还有更多的机会聆听王教授的谆谆教诲,并从他独立自主的人格魅力中受到鼓舞。这段在美国麻省剑桥城的时光也成为我们人生中最宝贵的经历[图1(c)~(e)]。以下部分将着重介绍我们在王教授指导下合作参与新加坡-麻省理工联盟项目所开展的几项酶技术领域的研究工作。

图1 王义翘教授与本文部分作者在新加坡-麻省理工联盟项目实施期间的合影Fig.1 Photos of Prof.Wang together with some of authors during the Singapore-MIT Alliance Program
酶的固定化是指通过物理或化学法将酶紧密地结合于非均相的载体上[26-27],它可以增加酶的稳定性,并通过重复使用降低酶的使用成本。固定于传统载体(厘米或毫米级)上的酶常常受限于较低的非均相催化效率和传质(例如王教授早期固定化甲醇氧化酶的工作[10]),而纳米级的载体有着更高的表面积/体积比,从而赋予固定化酶类似于均相催化的高效反应速率,但更小的载体通常意味着更困难的反应后分离。为了解决此矛盾,王教授启发性地提出了是否可以利用额外的作用力帮助分离纳米级的固定化酶。考虑到当时已经开发了许多基于四氧化三铁(Fe3O4)的磁性纳米颗粒(iron oxide magnetic nanoparticles),但鲜少用于酶的固定化,于是我们决定借助磁力来分离纳米级的固定化酶。王文合成了聚合物包埋铁氧化物核心的磁性纳米颗粒,并通过共价键将酶固定于其表面,从而开发了高活力可回收利用的纳米生物催化剂[图2(a)][28]。以不对称氧化苯硫基甲烷(thioanisole)作为模式反应,发现固定于磁性纳米颗粒上的氯过氧化物酶(chloroperoxidase)和游离的氯过氧化物酶有着几乎相同的催化活力和Km值,及同样完美的手性选择性。并且该磁性纳米生物催化剂可以通过磁场分离从而回收利用,实验结果表明该纳米催化剂在成功回收利用12 个循环后仍能保持原有活力。在后续课题中,王文构建了表面含有次氮基三乙酸镍(Ni-NTA)的磁性纳米颗粒,并利用它直接从细胞裂解液中一步纯化和固定含有组氨酸标签的重组酶[29]。在不对称水解对氯苯基环氧乙烷[2-(4-chlorophenyl)-oxirane]的模式反应中,相比于游离的环氧水解酶,固定在磁性纳米颗粒上的环氧水解酶保留了大部分的活力(80%)和相同的手性选择性,并可以通过磁场分离回收利用8 个循环(保持80%以上的产率)。基于这两项开创性的工作,我们课题组进一步开发了类似系统来固定化醇脱氢酶用于还原酮[30]、固定化脂肪酶用于生产生物柴油[31]、固定化乙酰高丝氨酸巯解酶(O-acetylserine sulfhydrylase)用于生产非天然氨基酸等[32]。这些成果的取得离不开项目最早期王教授开放型思维和交叉学科思想的启迪,通过引进先进的纳米技术应用于酶技术领域,创建了更为高效且可回收利用的纳米生物催化剂。
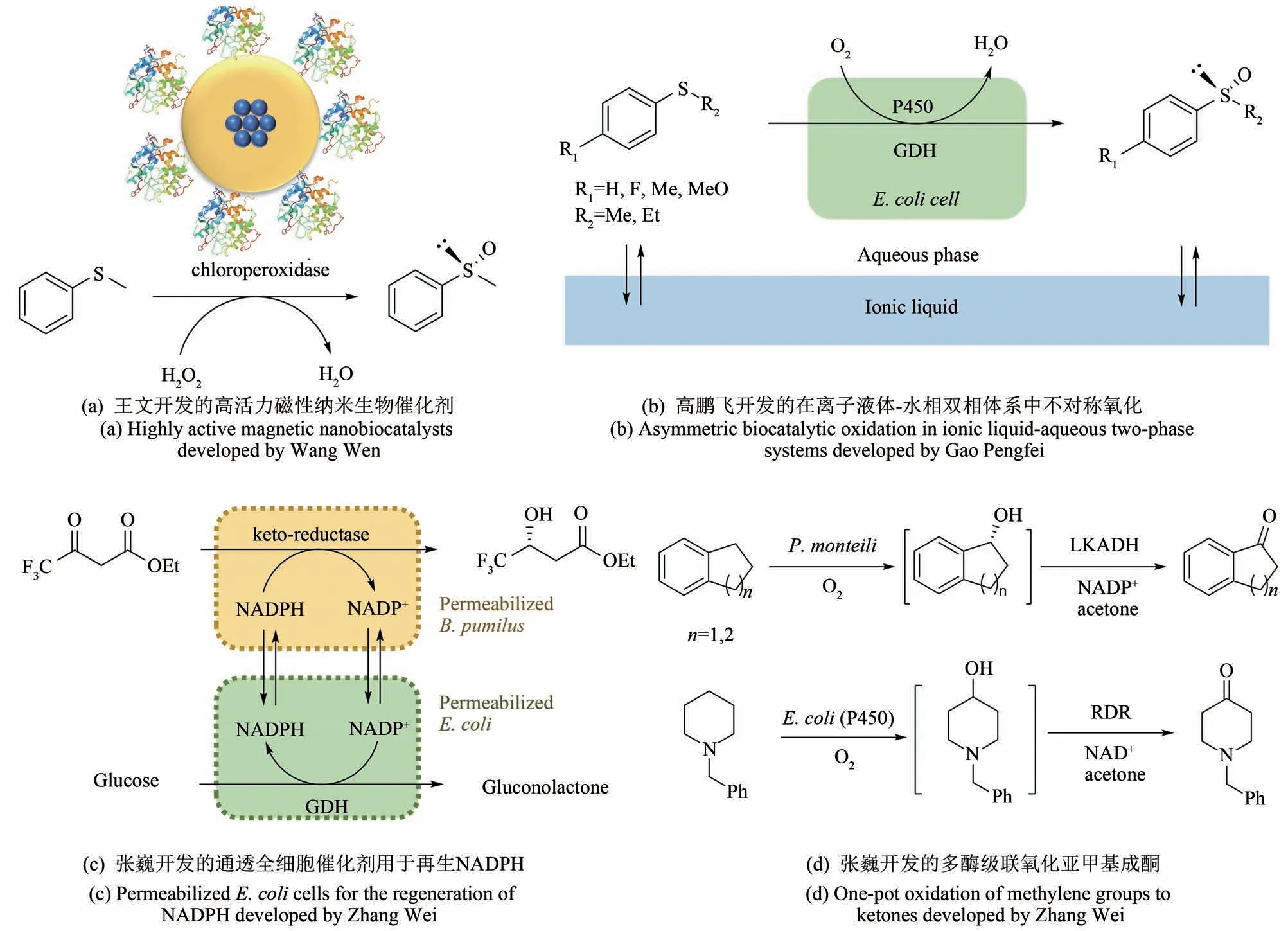
图2 王义翘教授在新加坡-麻省理工联盟项目里联合指导本文作者的四项酶技术领域的工作Fig.2 Four enzyme technology projects co-supervised by Prof.Wang through the Singapore-MIT Alliance Program
在非水溶剂中的酶促反应也一直是酶技术领域的研究热点,王教授在20 世纪90 年代就已经研究了反胶束体系中的酶促反应[15],除了传统的有机溶剂,近年来的研究热点主要围绕着更加绿色环保和生物相适性更好的离子液体(ionic liquid)[33]和低共熔溶剂(deep-eutectic solvents)[34]等。当时我们项目的一个课题旨在利用来自鞘氨醇单胞菌(Sphingomonassp.HXN-200)的P450pyr 单加氧酶催化不对称氧化苯基硫醚(thioanisole)合成手性苯基亚砜[Phenyl sulfoxide,图2(b)][35]。初步筛选实验室中不同的P450pyr 突变体后发现,在单一的水相(磷酸盐缓冲液)条件下,手性选择性最高的突变体生成几个苯基亚砜的ee值也只有80%~88%。通常提高手性选择性的思路是通过酶的定向进化来实现的[36],我们之前也已经成功定向进化了P450pyr 用于高选择性的羟化反应[37-39],但对于亚砜化反应,苦于没有合适的高通量筛选方法而止步。王教授于是建议尝试一些非水溶剂或双液相反应。鉴于很多氧化酶在非水溶剂中不稳定,且通过改变反应体系显著提高氧化酶的手性选择性还鲜有报道,刚开始我们并不愿尝试。在其他方案无果的情况下,高鹏飞筛选了不同的离子液体作为双液相的第二相,并惊喜地发现离子液体[P6,6,6,14][NTf2]显著提高了P450pyr 氧化反应的手性选择性,三个苯基亚砜产物的ee 值达96%~99%[35]。同时,该离子液体具有非常好的生物相适性,并且对苯基硫醚(底物)和苯基亚砜(产物)具有较高的溶解度,从而降低了底物和产物对P450pyr 酶的抑制作用。通过进一步研究后发现,该离子液体很有可能是通过降低水相中实际苯基硫醚的浓度,从而提高了P450pyr 的手性选择性。回过头来反思,王教授鼓励大胆尝试,打破常规性思维是该课题最终获得成功的重要因素。
酶催化过程经常消耗化学当量的辅因子,因此辅因子再生是众多酶催化过程中不可避免的一个重要环节[40]。作为行业先驱,王教授同早期的几个合作者也开发了ATP 的再生技术[12]。对于辅酶NAD(P)+/NAD(P)H 的再生,通常的思路是在大肠杆菌中共表达相应的辅酶再生的酶,或者利用分离纯化的酶来实现的。但这种方法并不适用于基于野生菌中尚未解析出相应功能的酶的情况,例如我们之前分离到一株短小芽孢杆菌(Bacillus pumilus)用于合成贝氟沙通(befloxatone)的手性中间体[41],但其中基于NADPH 酮还原酶尚不明确。在王教授的联合指导下,张巍开发了过表达葡萄糖脱氢酶(glucose dehydrogenase)的大肠杆菌作为通透的全细胞(permeabilized cells)用于再生NADPH[图2(c)][42]。将大肠杆菌和野生型短小芽孢杆菌组合,实现了高效的不对称酮还原。利用这个通透的全细胞辅酶NADPH 再生系统,其NADPH 的总转化数(total turnover number,TTN)达到了4200。并且这种通透的全细胞辅酶再生系统比利用纯化的酶有更低的成本,可作为常用的辅因子再生策略的重要补充。
酶作为具有催化功能的元件,可以通过理性设计和组装多个催化元件形成复合的催化系统,实现单一元件无法实现的化学转化的功能。这种多酶级联催化(enzyme cascade catalysis)可以实现一锅多步反应直接生产最终产物,避免了分离纯化中间产物的步骤,节省了人力和物力投入的同时还减少了废弃物的产生[43-49]。值得一提的是,在存在热力学上平衡反应的时候,级联的多步反应可以打破反应平衡,提高最终产物的产率。具体实施上,级联催化的多个酶可以首先通过分别表达纯化(甚至固定化),进而再混合在一起在容器中反应(无细胞酶催化合成),或者将多个酶表达在一个或多个重组微生物中构建独立的人工反应途径(全细胞酶催化合成)。王教授在20世纪70年代就利用部分纯化的非核糖体多肽合成酶(2 个组分),首次实现无细胞多酶制备级全合成短杆菌肽S[11]。这个早期的工作利用的是天然的生物合成途径,而通过人工设计非天然的酶催化路线,利用不同来源的酶,可以创造出新的化学转化。在王教授的联合指导下,张巍结合单加氧酶催化的羟基化反应和醇脱氢酶催化的醇氧化反应实现了一锅法转化亚甲基氧化成酮[图2(d)][50]。第一个具体的反应结合了一株含有单加氧酶的蒙氏假单胞菌(Pseudomonas monteilii)和商业化的醇脱氢酶LkADH(开菲尔乳杆菌来源),用以氧化四氢化萘(tetralin)生成1-四氢萘酮(1-tetralone,得率达88%)以及二氢茚(indan)生成1-二氢茚酮(1-indanone,得率达87%)。另一个反应结合了表达P450pyr的大肠杆菌和纯化的醇脱氢酶RDR,用以氧化N-苄基哌啶(N-benzyl-piperidine)生成N-苄基-4-哌啶酮(N-benzyl-4-piperidone,得率达86%)。在以上脱氢酶催化的醇氧化反应中,所需的辅因子NAD+和NADP+是通过加入丙酮得以再生的(总转化数达4000以上)。由于羟化酶具有极高的位置选择性,在反应中并未检测到其他位置氧化的副产物的生成。
鉴于上述反应较为简单且产物不具手性,接下来我们开发的另一个双酶级联反应,环氧化-水解生产手性二醇[51],被王教授尖锐地指出有机化学也可以实现同样的反应,且产物的复杂度远不如他早期开发的短杆菌肽S。在王教授的鞭策和激励下,我们决定利用合成生物学的工程化和模块化思想,由下至上地构建独立于细胞代谢的反应途径,实现更为复杂的、化学法无法企及的一锅反应,从末端烯烃一锅法合成手性α-羟基酸、邻氨醇和α-氨基酸(图3)[52]。吴淑可首先利用合成生物学的模块化思想,将两个酶催化反应(及其辅助反应)组成一个模块,根据总反应历程共设计了4 个酶催化模块,相应模块的具体职能如下:模块I,环氧化酶-环氧水解酶催化烯烃转化成邻二醇;模块II,醇脱氢酶-醛脱氢酶催化邻二醇氧化成α-羟基酸;模块III,醇脱氢酶-ω-转氨酶-丙氨酸脱氢酶催化邻二醇转化成邻氨醇;模块IV,羟基酸氧化酶-过氧化氢酶-α-转氨酶-谷氨酸脱氢酶催化α-羟基酸转化成α-氨基酸。这样通过组合不同的模块足以实现目标中所设计的三类烯烃转化。以苯乙烯合成手性扁桃酸(mandelic acid)、苯乙氨醇(phenylethanolamine)、苯甘氨酸(phenylglycine)为模式反应,我们从多种微生物中筛选并克隆了相应的酶,并将每个模块的基因分别构建到4个兼容的质粒上,组合后转入大肠杆菌,从而最终筛选出了3 株最优的菌株来分别催化苯乙烯生产S-扁桃酸、S-苯乙氨醇和S-苯甘氨酸(转化率为86%~95%,分离得率为60%~72%)。与此同时,该多酶级联催化还可以转化多个不同取代的苯乙烯生成相应的取代产物。由于多个酶的高位置和手性选择性,所有最终手性产物的ee 值达91%~99%。从简单易得的烯烃直接一锅法制备高值的手性精细化学品,这3种多酶级联催化反应全部在温和的条件下进行,并且只消耗便宜且环保的葡萄糖、氧气和氨作为计量的反应物,为绿色生物制造拓展了新的版图。此外,这三条途径涉及4~6 步线性反应,需要4~8 个不同来源的酶参与,在大肠杆菌中构建出了独立于细胞代谢的人工途径,证明了较多步骤的酶级联催化的高效性。总结成果之余,我们深刻地感受到该课题最终成功开发与王教授对本项目早期的指导和激励是密不可分的。
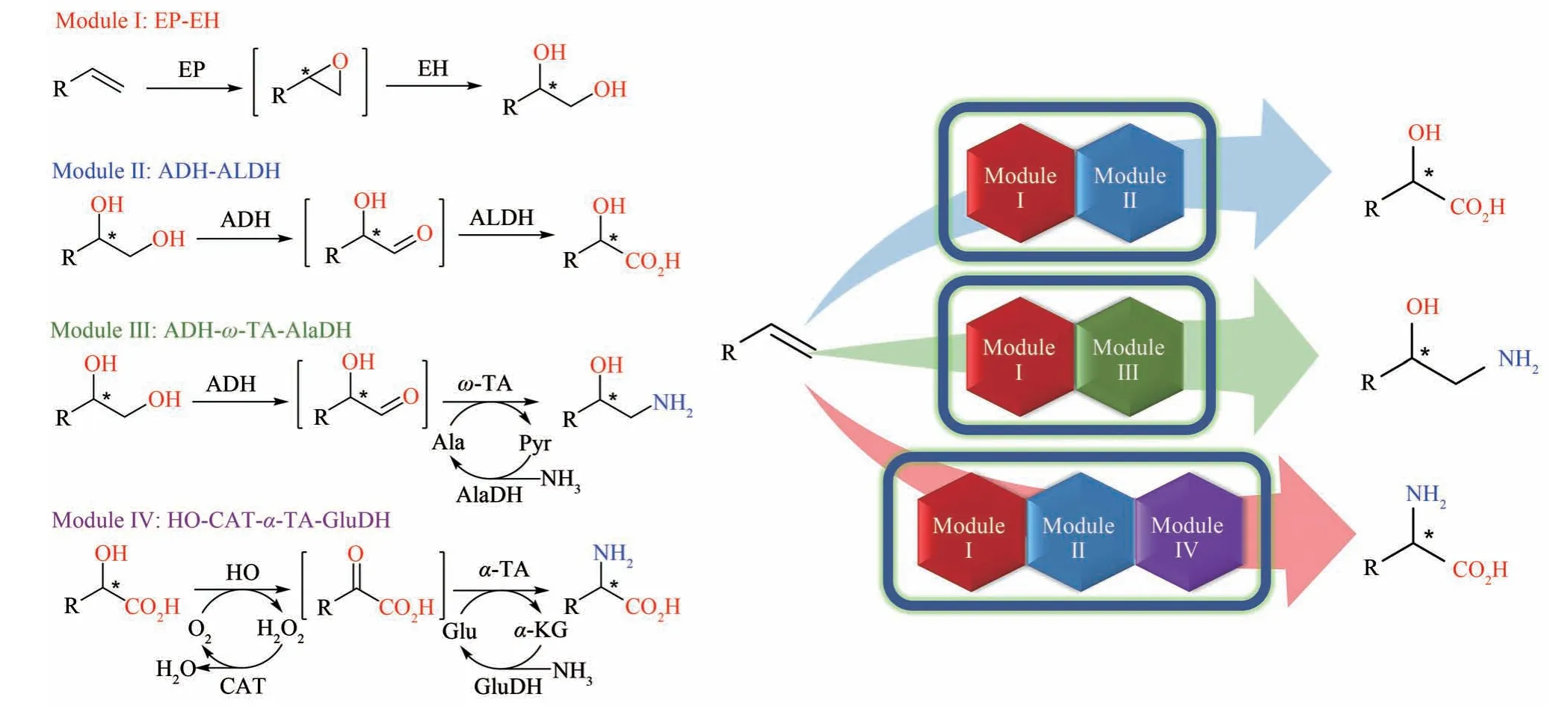
图3 王义翘教授在新加坡-麻省理工联盟项目里联合指导本文作者吴淑可开发的模块化多酶级联催化转化烯烃生成三类手性分子EP—环氧化酶;EH—环氧水解酶;ADH—醇脱氢酶;ALDH—醛脱氢酶;ω-TA—ω-转氨酶;AlaDH—丙氨酸脱氢酶;HO—羟基酸氧化酶;α-TA—α-转氨酶;CAT—过氧化氢酶;GluDH—谷氨酸脱氢酶Fig.3 Modular multi-enzyme cascade catalysis for the transformation of alkenes into three types of chiral molecules developed by Wu Shuke and co-supervised by Prof.Wang through the Singapore-MIT Alliance.EP—epoxidase;EH—epoxide hydrolase;ADH—alcohol dehydrogenase;ALDH—aldehyde dehydrogenase;ω-TA—ω-transaminase;AlaDH—alanine dehydrogenase;HO—hydroxy acid oxidase;α-TA—α-transaminase;CAT—catalase;GluDH—glutamate dehydrogenase
在此项目的基础上我们和其他课题组还进一步扩展了烯烃的多酶级联催化,用于生产更多不同种类的手性或大宗芳香族化学品,例如利用来自施氏假单胞菌(Pseudomonas stutzeri)的特别的D-转氨酶生产重要的抗生素侧链D-苯甘氨酸[53];利用苯乙烯天然降解途径中的氧化苯乙烯歧化酶(styrene oxide isomerase)生产苯乙酸、苯乙醇和苯乙胺[54-55];利用扁桃酸天然降解途径中的脱羧酶进一步生产苯甲胺和苯甲酸[56-57];利用来自枯草芽孢杆菌的丁二醇脱氢酶和不同的ω-转氨酶生产R-苯甘氨醇或S-苯甘氨醇[58-59]等。考虑到烯烃大都是基于不可再生的化石燃料的,为了直接利用可再生的生物基原料,我们设计了另外一个酶催化模块,包括拟南芥来源的氨基裂解酶(phenylalanine ammonia lyase)和黑曲霉来源的脱羧酶(phenylacrylic acid decarboxylase)来实现从生物基苯丙氨酸生产苯乙烯。将该酶模块和上文已经开发出的酶模块有机结合之后,实现了从生物基苯丙氨酸出发的多酶级联催化生产多种非天然手性化学品[60]。为了直接利用更为廉价的生物基原料,我们改造了一株大肠杆菌以葡萄糖或甘油发酵生产苯丙氨酸,并开发了“偶联发酵-生物转化技术”(coupled fermentationbiotransformation)原位转化发酵液中的苯丙氨酸用以高效生产具有抑菌作用的大宗芳香族化学品苯甲酸(防腐剂)和苯乙醇(香料)[57,61]。以上这些工作展示了多酶级联催化的许多优点,例如节省了分离纯化中间产物的步骤,不易受反应中间体或最终产物的抑制等等。近年来,来自世界各地的研究者也开发了许多新型高效的多酶级联催化反应用于生产大宗化学品、手性化学品、复杂的人工药物和天然产物等[43-49],我们将继续重点介绍近年来所开发的生产手性和大宗化学品的多酶级联催化反应。
3 多酶级联催化的最新进展
手性胺是一类极为重要的手性合成中间体,被广泛应用在合成药品、精细化学品、农用化学品的生产和制备中[62]。相较于化学法,酶催化合成手性胺通常具有更高的选择性,且反应条件温和,反应体系绿色无毒,其中最著名的范例是美国Codexis 公司定向进化ω-转氨酶合成西他列汀[63]。通常酶催化合成手性胺需要利用酮作为底物,而非手性或消旋的醇是更为廉价易得的底物,但尚且没有单一的酶可以直接转化醇变为胺。英国曼彻斯特大学的Turner教授开发了一个简单的“借氢”双酶级联催化,利用醇脱氢酶(ADH)和人工改造的胺脱氢酶(AmDH,系王义翘教授的学生Bommarius教授开发[64]),直接转化一系列芳香族或脂肪族的二级醇生成相应的手性胺,而无需消耗化学计量的氧化/还原剂[图4(a)][65]。该双酶级联反应只需要氨水作为化学计量的试剂,且大部分产物手性胺都达到了光学纯。但醇脱氢酶的高手性选择性亦是一把双刃剑,例如短乳杆菌来源的脱氢酶只能氧化R型的芳香醇,当需要利用廉价易得的消旋醇作为底物的时候,必须利用两个手性选择性互补的醇脱氢酶。几乎同时,华东理工大学许建和课题组也报道了类似的双酶级联催化从脂肪族的醇合成手性胺[66]。他们利用一个天蓝色链霉菌来源的羰基还原酶(ScCR)和改造后的胺脱氢酶,转化了一系列脂肪族的二级醇和1-苯乙醇生成相应的手性胺。最近他们还增加了一个P450 酶扩展了该双酶级联反应,直接从环己烷和乙苯分别生产环己胺和R型1-苯乙胺[67]。除了改造的胺脱氢酶,ω-转氨酶也可以和醇脱氢酶组合用于转化醇生成胺,但是需要额外的较为复杂的氨基供体及其再生系统(例如L-丙氨酸和丙氨酸脱氢酶),且最终手性胺的转化率往往不高[68]。最近,我们课题组设计了一个简单的醇脱氢酶-ω-转氨酶双酶级联催化体系,实现了消旋的二级醇到手性胺的高效转化[图4(b)][69]。其关键点是对近平滑假丝酵母来源的S-选择性的醇脱氢酶(CpSADH)进行了定向进化,使其对S型和R型的二级醇具备相近的活力,使得只需使用单个酶(CpSADH-W286A)就可以充分氧化一系列消旋的二级醇。该体系利用异丙胺作为转氨酶的氨基供体,其副产物丙酮刚好可以被醇脱氢酶还原以再生NAD+。这样,该体系只需要两个酶和消耗廉价的化学当量异丙胺,就可以高效转化消旋的二级醇生产手性胺。上述这些例子主要是生产手性一级胺,而二、三级胺无法利用ω-转氨酶或一般的胺脱氢酶直接制备。最近Turner 课题组率先利用最新发现源于米曲霉的还原胺化酶(催化羰基和一级胺生成亚胺并且还原为二级胺)[70],进一步结合几个不同的醇脱氢酶或醇氧化酶构建了双酶级联催化体系,实现了从一系列不同的一、二级醇和一级胺直接合成相对应的二级胺[71]。
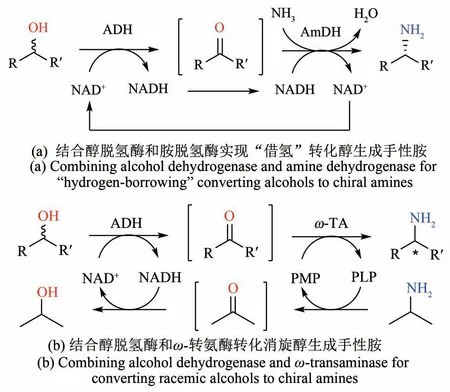
图4 多酶级联催化转化二级醇一锅法生产手性胺Fig.4 Multi-enzyme cascades for converting secondary alco‑hols into chiral amines
与手性胺极为相关的是手性氨基酸,不仅编码蛋白的19 种天然氨基酸是手性的,许多手性的非蛋白氨基酸也是多种生物活性物质的重要组成部分,例如内酰胺类抗生素、非核糖体多肽类天然产物、人工合成的多肽类药物等[72-73]。鉴于手性氨基酸的重要性,一系列多酶级联催化过程已经进行了充分开发用于制备手性α-氨基酸,例如早期利用海因酶-氨甲酰水解酶-消旋酶(海因酶法)制备D-氨基酸[74],以及最近通过类似于醇转化成胺的级联反应利用双酶级联催化反应可以转化α-羟基酸生成手性α-氨基酸等[75]。大部分早期开发的酶级联催化反应只涉及简单的官能团转换,并没有涉及更为复杂的“碳—碳”键的生成。最近江南大学刘立明课题组基于合成生物学模块化的思想开发了一种多酶级联催化反应,通过“引入-重置-再引入”手性基团的策略,实现了一系列手性α-羟基酸和α-氨基酸的合成[图5(a)][76]。其中的模块化元件涉及两部分,第一部分是基础模块,利用一个来自铜绿假单胞菌的苏氨酸醛缩酶(threonine aldolase)和来自棒杆菌的苏氨酸脱氨酶(threonine deaminase)分别催化的醛缩反应和非典型的脱氨反应,转化醛和甘氨酸生成α-酮酸。第二部分是扩展模块,利用羟基异己酸脱氢酶(hydroxyisocaproate dehydrogenase)或氨基酸脱氢酶(amino acid dehydrogenase)及其辅助酶重新引入手性羟基或氨基,将α-酮酸转化为手性α-羟基酸或α-氨基酸。通过将相应模块的基因分别构建到兼容的质粒上,并组合转入大肠杆菌,从而筛选出最优的菌株用于催化相对应的整个级联反应,最终实现从不同的醛和甘氨酸出发合成了高手性纯度的S型和R型的α-羟基酸和α-氨基酸。尽管产物浓度(约1 g/L)尚待提高,该多酶级联催化体系充分利用了合成生物学模块化的思路,显著地拓展了绿色合成手性α-羟基酸和α-氨基酸的方法。另一类重要的手性氨基酸是β-氨基酸。目前通过酶催化合成β-氨基酸的思路主要有两条。一种是利用氨基裂解酶在α,β-不饱和羧酸上经由高位置和手性选择性地加上氨基,例如微生物所的吴边课题组利用计算机设计改造了天冬氨酸酶(aspartase)合成了一系列手性β-氨基酸[77],江南大学刘立明课题组利用酪氨酸酚裂解酶(tyrosine phenol lyase)和酪氨酸氨基变位酶(tyrosine aminomutase)从苯酚和丙酮酸合成了R型β-酪氨酸[78]。另一种合成β-氨基酸的策略是利用转氨酶或者脱氢酶转化β-酮酸,但是由于β-酮酸不稳定,通常需要外加一个酶用于生成β-酮酸并进行原位转化,从而通过双酶级联催化合成手性β-氨基酸。例如天津工业生物技术研究所朱敦明课题组结合β-氨基酸脱氢酶与脂肪酶或腈水解酶,从β-酮酸酯或β-酮腈一锅法合成手性β-氨基酸[图5(b)][79]。
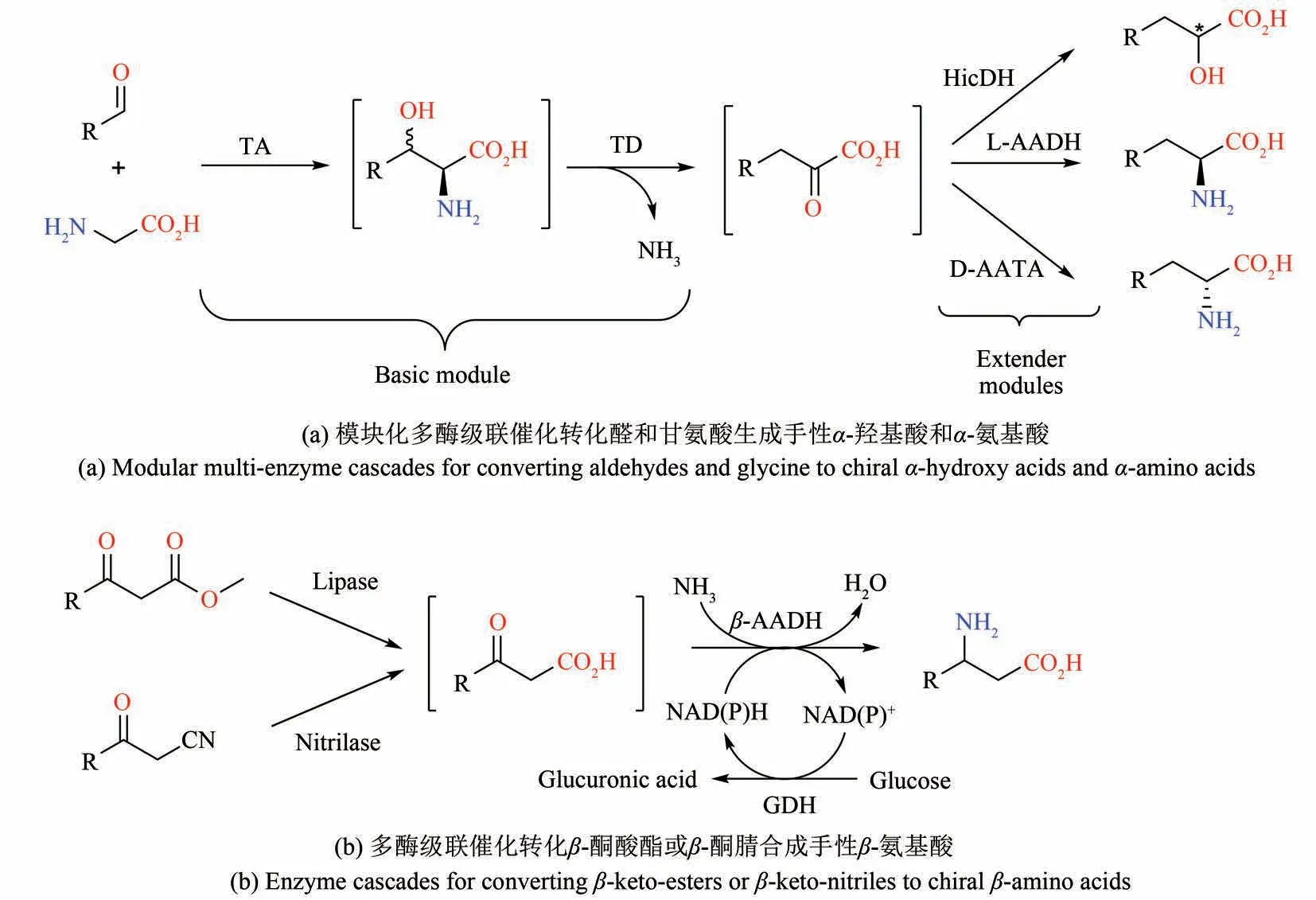
图5 多酶级联催化生产手性α-氨基酸和β-氨基酸Fig.5 Multi-enzyme cascades for the production of chiral α-amino acids and β-amino acids
除了高附加值的手性化学品,多酶级联催化也可以应用于合成各种大宗化学品,特别是广泛应用的聚合物的单体等[80]。浙江大学叶丽丹等[81]报道了由P450 酶、醇脱氢酶、ω-转氨酶组成的多酶级联催化转化月桂酸(十二碳酸)生成尼龙12 单体(ω-氨基十二烷酸)。值得一提的是,他们不仅表达了几个辅助酶用于辅酶和氨基供体再生,而且还根据代谢工程的思路敲除了宿主细胞里降解的β-氧化途径中的相关酶以及过表达转运通道蛋白用于辅助底物转运。对于更广泛应用的中链二元酸,湖北大学李爱涛团队设计了一个6步的多酶级联催化体系一锅转化环烷烃(C5~C8)生产中链二元酸(C5~C8),其中相应的催化职能依次包括P450 催化羟化、醇脱氢酶氧化醇、拜尔维利格单加氧酶(Baeyer-Villiger monooxygenase)氧化酮、内酯酶(lactonase)水解、醇脱氢酶氧化羟基酸、醛脱氢酶氧化醛酸[图6(a)][82]。该系统同样采用了合成生物学模块化的思路,且每个模块包含辅助酶用于辅酶再生,但与之前不同的是,他们将每个模块单独构建在一株大肠杆菌中,利用重组大肠杆菌菌群(consortia)的催化策略实现一锅合成。这种菌群催化的策略比利用单一重组菌更容易调控优化,在最优的条件下,重组菌群可以从环己烷或环己醇一锅法合成约5~8 g/L的己二酸。利用相同的策略,改用羧酸还原酶和酮还原酶,他们还开发了另一个相似的多酶级联催化合成1,6-己二醇[83]。另一个最新的工作是来自韩国建国大学的Yun教授课题组,他们设计了一个多酶级联反应从环烷胺(C4~C6)合成尼龙单体内酰胺(C4~C6),其独特之处在于其中的ω-转氨酶可以利用环烷胺中的氨基合成ω-氨基酸,从而无需额外的氨基供体[图6(b)][84]。在以上大部分的工作中,最终产物的浓度尚比较低(<10 g/L),而为了在工业上与现有的化学法合成形成竞争力,大宗化学品的酶催化合成需要极高的效率(时空产率和转化率)。现如今,酶的大规模挖掘[85]、理性设计、定向进化手段[86]和基于合成生物学的微生物调控手段[87]可以极大地提升(微生物体内)多酶级联催化的效率。例如凯赛生物基于微生物内的多酶级联氧化长链烷烃,大规模商业化生产长链二元酸,已成为多酶级联催化生产大宗化学品的典范。
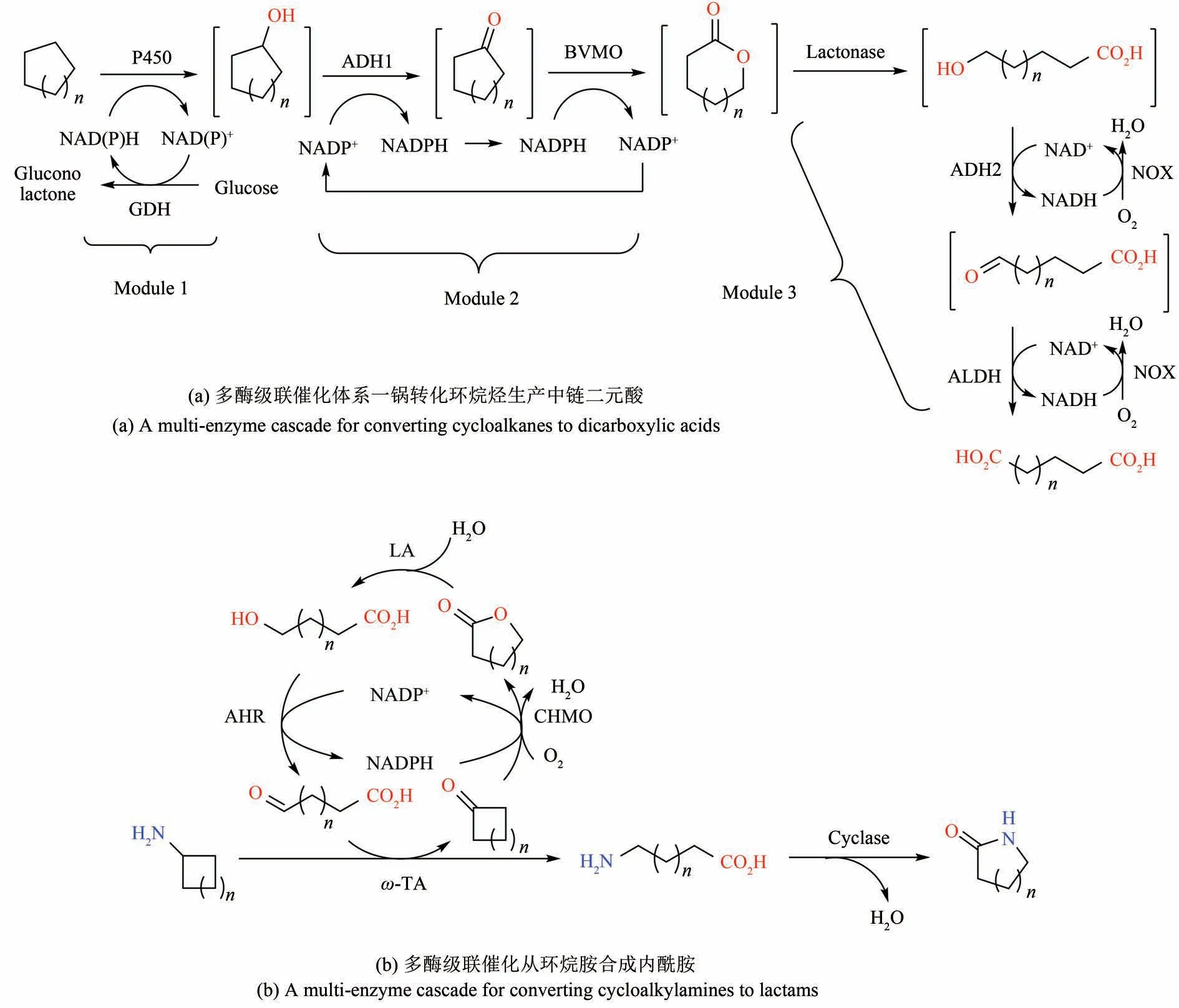
图6 多酶级联催化生产大宗化学品Fig.6 Multi-enzyme cascades for the production of bulk chemicals
此外,近年来在合成复杂的天然产物和人工药物等领域,多酶级联催化策略也被越来越广泛地采用[24,88],例如美国默克公司利用一锅9 酶3步法合成新型抗艾滋病药物Islatravir[89],相关进展也于近期被很好地总结评述[24],这里就不再赘述。
4 多酶级联催化的发展方向以及王义翘教授工作的启示
类似于合成生物学的发展[2],创造性地结合其他学科也是多酶级联催化发展的重要方向。在与化学学科的交叉方面,进一步结合各种小分子催化、金属催化、光催化、电催化剂组成化学-酶级联催化体系可以进一步扩展一锅合成体系,实现单独化学催化剂或者生物催化剂无法实现的功能[90-91]。例如美国伊利诺伊大学赵惠民组与加州大学伯克利分校Hartwig 组结合光催化的烯烃顺反异构和烯酮还原酶实现的不对称还原反应[92],瑞士巴塞尔大学Ward 组结合多酶级联反应和金属催化的烯烃复分解实现从生物基的油酸合成大宗化学品环烯烃[93],荷兰代夫特理工Hollmann 组与韩国Park 组利用油酸水合酶与光活化的脱羧酶从不饱和脂肪酸合成手性脂肪醇等[94]。此外,许多天然的多酶催化体系也给人工多酶体系的发展提供了灵感,例如多个酶在空间上有序地组织可以进一步提高级联催化的效率[95],武汉大学刘天罡组与香港中文大学夏江组,利用一对多肽标签实现了两种酶的有效组装,提高了在体外或细胞内的级联催化效率[96]。除了在空间上的优化,利用不同温度也可以极大地优化多酶催化体系,例如上海交通大学许平组利用嗜热菌来源的多种酶,开发了热稳定的双酶级联反应从阿魏酸合成香草醛,避免了产物的降解和副产物的生成[97-98]。
以上涵盖了多酶级联催化未来的重要发展方向,而当我们再次回顾王义翘教授早期在酶技术领域的工作,相关的工程学研究思路和跳出瓶颈的思维魄力,仍对本领域现今以及未来的发展有着重要的启示和指引。王教授早在40 多年前就开始研究酶催化转化甲醇,如今利用碳一底物(例如二氧化碳、甲烷、甲醇等)依旧是酶催化与微生物炼制的研究热点[21-23]。例如德国马普所Erb 组设计并开发了一条全新的固定二氧化碳的17 个酶的级联催化循环途径,在体外优化的条件下比天然的卡尔文循环固碳效率要高[99];天津工业生物技术研究所的江会锋组结合乙醇醛合酶、乙酰磷酸合酶和磷酸转乙酰酶构建了一条全新的从甲醛合成乙酰辅酶A 的途径[100]。由于越来越多转化碳一底物的酶被发现、开发和人工进化,我们可以预期许多转化碳一底物的多酶级联催化将会被陆续开发出来,势必将极大地扩展体外和体内转化碳一原料的生物合成系统。而在另外一个方向上,本文作者和王教授也一起开发了高活力、高稳定性、可回收利用的磁性纳米生物催化剂,大大降低了单个酶催化的成本。如果将多个酶高效有序地固定在纳米级的载体上,不仅能提高多酶级联催化的效率,还能通过多次回收利用降低多酶级联催化的成本。最后一点,也是非常重要的一点,定量思想和工程化思想常常贯穿在王教授多个早期的酶技术项目,相关的研究并不满足于浅尝辄止,而是更深入地研究了酶催化反应动力学并建立了对应的数学模型等。然而近年来,纵观多酶级联催化的进展,往往偏重于优化反应体系和最终产物的合成制备,而忽视了对多酶催化过程的定量分析和动力学刻画。我们应该看到,对体外或细胞内多酶级联催化过程的数学建模和定量分析描述,将为进一步提高多酶反应的效率提供理论基础和优化指导,从而帮助提高多酶级联催化的效率接近或达到工业化应用的标准。这也是将这种基于自下而上的思路所构建的多酶级联催化反应,真正地提高到合成生物学的理论高度所必须践行的标准。
