S空位调节扶手型二硫化钼纳米带的电子性质
2021-08-16高云婕周小羽
高云婕,高 见,周小羽
(1.扬州科技学院电子工程学院,扬州 225009;2.扬州大学物理科学与技术学院,扬州 225002)
1 引 言
过渡金属硫属化合物(TMDs),由于优良的物理化学性质引起了人们的广泛关注[1-5].其中,二硫化钼作为典型代表,其二维体系具有类石墨烯的蜂窝状点阵结构特点,单层二硫化钼由SMo-S组成,其中一个Mo层封闭在两个S层中,原子正六边形排列,层间通过范德华力连在一起[6,7].体相MoS2是带隙为1.3 eV的间接带隙半导体,而单层MoS2是带隙为1.8 eV的直接带隙半导体[8,9],能更好地吸收可见光.
低维MoS2受量子束缚效应的作用,展现出奇特的光、电、磁及催化性质,在纳米器件、纳米新能源、纳米催化等领域具有潜在的实际应用价值,已经成为当今纳米材料研究的前沿和热点.对于二维MoS2沿纵向切割时可形成准一维MoS2纳米带(MoS2NRs),根据其边界结构特点,可形成扶手椅型纳米带(AMoS2NRs)和锯齿型纳米带(ZMoS2NRs),但两者表现出了完全不同的电、磁性质,锯齿状MoS2纳米带(ZMoS2NRs)是具有铁磁性(FM)边缘态的磁性金属[10],而扶手椅型MoS2纳米带(AMoS2NRs)表现为非磁性半导体,其带隙随纳米带宽度增加而增加,并从间接带隙转变为直接带隙[11].说明MoS2纳米带电、磁、输运等性质依赖于边缘态[1,2].然而在样品的制备过程中,缺陷不可避免,其中S空位最为典型.潘等人发现,对有空位的ZMoS2NRs边缘进行修饰时,能够使其从金属性质变为半金属性质[12].所谓半金属材料是指在一个自旋通道上表现出金属特性,在另一个自旋通道中表现出绝缘体性质.半金属材料被认为是未来纳米器件中提供100%自旋极化电流的理想自旋电子材料[13-17].事实上,AMoS2NRs中的原子空位缺陷很容易得到,甚至可以通过实验实现其浓度来控制[18-20].然而,关于空位对其电子性质的影响的报道极少,这里,我们采用密度泛函理论(DFT)方法,研究S空位(VS)对AMoS2NRs性质的影响.电子结构的计算和磁性分析表明,S空位的位置对体系物性影响比较大,当S空位在内部即a-g处时,AMoS2NRs均是非磁半导体,但当S空位在边缘即h处时,AMoS2NRs表现为磁性半金属.主要原因是边缘S空位能极大地改变体系的结构,特别是边缘的结构.进而打破了原有的平衡,使边缘电荷分布发生改变,从而改变了AMoS2NRs的电子结构,使其被调节成半金属.但随着其空位浓度时的增加,AMoS2NRs体系可被调节成稀磁半导体.这些有趣的发现将使得低维MoS2材料在自旋电子学上存在潜在的实用价值.
2 计算模型和方法
本计算,基于VASP软件(Vienna ab-initio simulation package)[21,22]的密度泛函理论(DFT),用Perde-Burke-Ernzerhof(PBE)的广义梯度近似(GGA)处理电子交换修正势[23],S-Mo-S层间的van der Waals相互作用由vdW(van der Waals)修正[24]来描述,用PBE+vdW方法对MoS2纳米带计算的可靠性已经被证实[25,26].平面波基组展开的截止能量使用500eV.所有的晶格常数和原子位置都进行了优化,达到其Hellman-Feynman力小于0.01 eV/Å,总能量收敛到1.0×10-5eV.在Monkhorst-Pack方案的基础上,采用特殊的k点,用1×11×1网格几何优化、1×21×1网格进行电子结构计算.计算得到的晶格常数为3.17Å,与实验值(3.16Å)[27]一致.
实验中,不同结构的MoS2纳米带可通过切割单层MoS2获得,如图1(a)给出了带宽N=8的AMoS2NRs,在计算中,沿纳米带的方向施加一维周期边界条件,另两个方向则施加大于10Å的真空层,以去除相邻模型之间的耦合作用.图1(b)给出了纯AMoS2NRs的能带和原子分波态密度图,很明显,自旋向上通道和自旋向下通道完全对称,AMoS2NRs是一个非磁性的半导体.为了考察S空位的位置对AMoS2NRs物性的影响,我们研究了S空位在不同位置处(如位点a、b、c、d、e、f、g和h)的情况,如图1(a)所示.

图1 (a):纯AMoS2 NRs的侧视图,N代表纳米带的带宽,紫色、黄色球分别代表MO、S原子,a~h字母代表位点;(b):带宽N=8的纯AMoS2 NRs的能带结构和原子分波态密度图;Fig.1 (a):Side view of pure AMoS2 NRs,N represents the bandwidth of the nanoribbons,The purple and yellow spheres represent Mo and S atoms respectively,letters a~h represent the site(b):Energy band structure and atomic partial state density of pure AMoS2 NRs with bandwidth N=8;
3 结果与讨论
首先研究S空位对AMoS2NRs结构的影响.对于纯AMoS2NRs,边缘的Mo-S平均键长为2.410Å,S空位在不同位置时,AMoS2NRs边缘Mo-S的平均键长为2.389Å,其中空位在边缘,即a和h位点时,Mo-S键长变化较大,分别是2.263Å和2.261Å.此外,S空位对Mo-Mo键也产生了一定影响,边缘的平均键角与纯AMoS2NRs的平均键角存在一定的差异,也就是说S空位对晶体结构产生了影响,尤其当空位出现在边缘时,AMoS2NRs结构变化最明显.由于AMoS2NRs的电磁性质强烈依赖于边缘的结构,空位使其边缘晶格发生了结构畸变,进而诱导AMoS2NRs的晶体场的变化,必将对其电子结构产生重大影响.
在研究其电子性质之前,我们首先需要考虑空位的出现对AMoS2NRs稳定性的影响,形成能是研究其稳定性的重要指标.形成能定义为=(Edefect+nμs)-Eperfact[10],这里Edefect和Eperfact分别是有S空位缺陷和没有S空位缺陷AMoS2NRs的总能量.n是完备的AMoS2NRs中移除的S原子数目,μS是S原子的化学势,这在很大程度上取决于其实验生长条件,因此需要分别考虑富Mo和富S条件.表1显示了AMoS2NRs的S空位在不同位置的形成能.在富Mo条件下,形成能在-3.31~-0.69 eV范围内,为负值,但在富S条件下,形成能为0.93~3.54 eV,为正值,说明在富Mo条件下,S空位较易形成.此外,S空位在边缘位置(a位和h位)的形成能低于内部位点(b、c、d、e、f和g),表明边缘位置很容易形成S空位,这是由于悬挂键的存在,边缘位置不稳定且活跃而导致的.
表1 S空位的位置、富S和富Mo条件下的形成能单位eV)、边缘位置Mo-S的键长,中间位置SS键长和带隙E g(eV).Table 1 position of S vacancy,formation energiesin eV)under rich S and rich Mo conditions,bond lengths(in A)of edge position Mo-S,bond lengths(in A)of middle position S-S and band gaps E g(in eV).
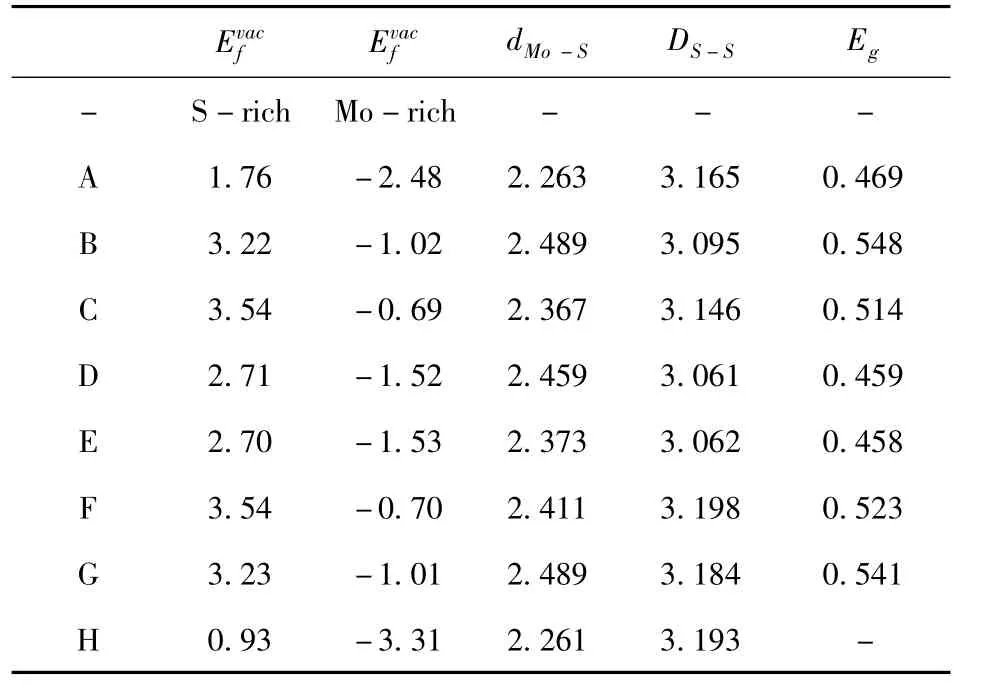
表1 S空位的位置、富S和富Mo条件下的形成能单位eV)、边缘位置Mo-S的键长,中间位置SS键长和带隙E g(eV).Table 1 position of S vacancy,formation energiesin eV)under rich S and rich Mo conditions,bond lengths(in A)of edge position Mo-S,bond lengths(in A)of middle position S-S and band gaps E g(in eV).
图2 给出了S空位不同位置时,扶手型二硫化钼纳米能带结构和分波态密度图(PDOS),我们发现,其电子结构图各不相同,其主要原因是S空位在一定程度上改变了二硫化钼纳米带的晶体结构,进而引起其能带结构的变化.当空位出现在中间位置时,能带图中显示,其自旋向上和自旋向下通道对称,AMoS2NRs都表现出非磁性半导体的性质.PDOS分析表明,费米面附近分波态密度主要来源于边缘的Mo-4d和S-3p轨道贡献.由于S空位对AMoS2NRs结构的影响,费米能级附近的电子态有所不同,因此,S空位在不同位置时,AMoS2NRs体系具有不同的带隙,即S空位在a,b,c,d,e,f,g位点时,带隙分别为0.469,0.548,0.514,0.459,0.458,0.523和0.541eV.
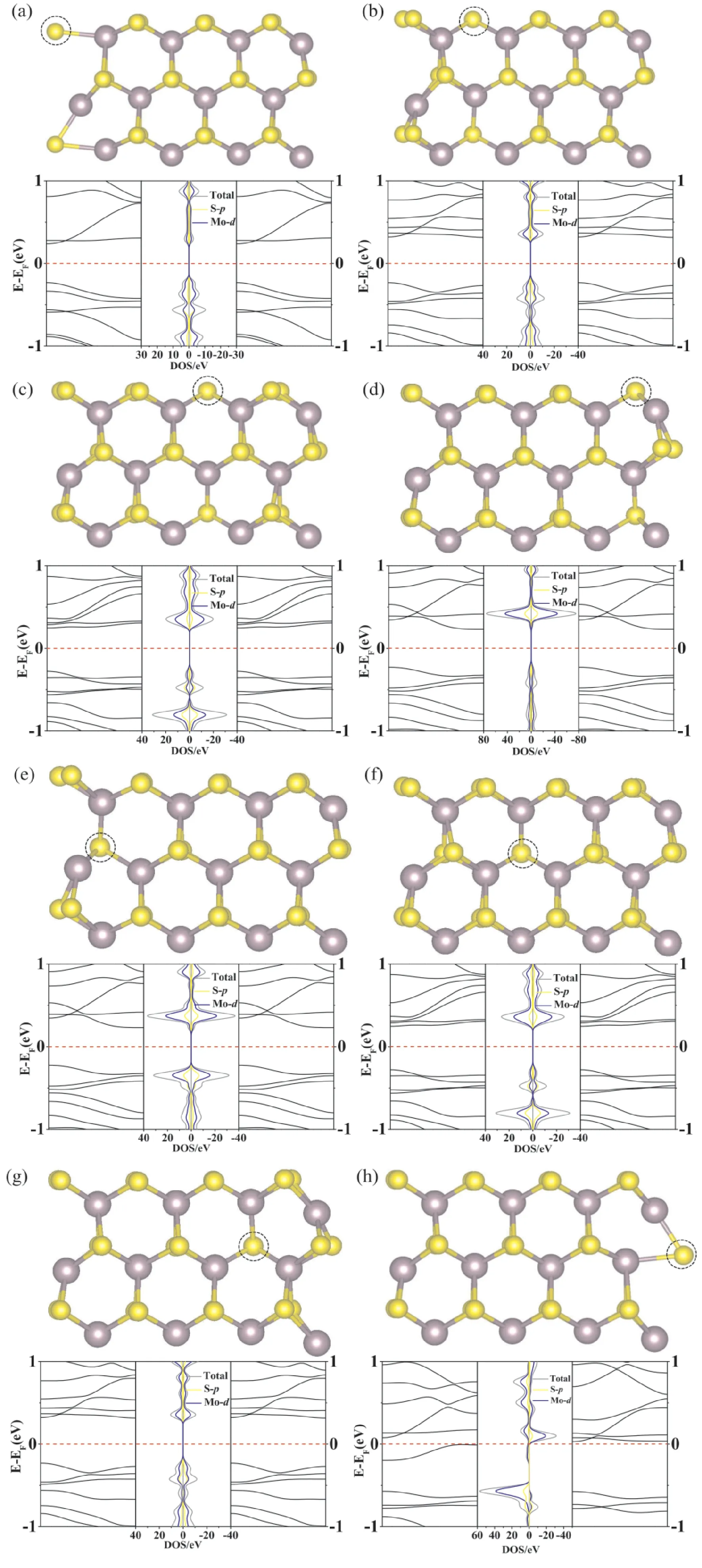
图2 S空位分别在a、b、c、d、e、f、g和h位点时AMoS2 NRs的能带结构和分波态密度图(PDOS).Fig.2 Energy band structures and partial state density(PDOS)diagrams of AMoS2 NRs with S vacancies at a,b,c,d,e,f,g and h sites,respectively.
有趣的是,当空位出现在边缘h位置时,AMoS2NRs的能带结构发生很大的变化,其性质从非磁半导体变成了磁性半金属态.如图2(h),从能带图和原子部分PDOS可以发现,自旋向上通道费米能附近有明显的能带穿过费米能,而在其自旋向下的费米能附近却没有能带穿过,这是半金属性质的重要特征.由图3(b)自旋密度图发现,空位出现在h位置时,AMoS2NRs右侧有明显的磁性,磁性主要来源是边缘的Mo,这也进一步表明AMoS2NRs的电、磁性质主要依赖于边缘,当空位出现在边缘时,边缘位置的晶体结构发生明显的畸变,这是磁性产生的主要原因.
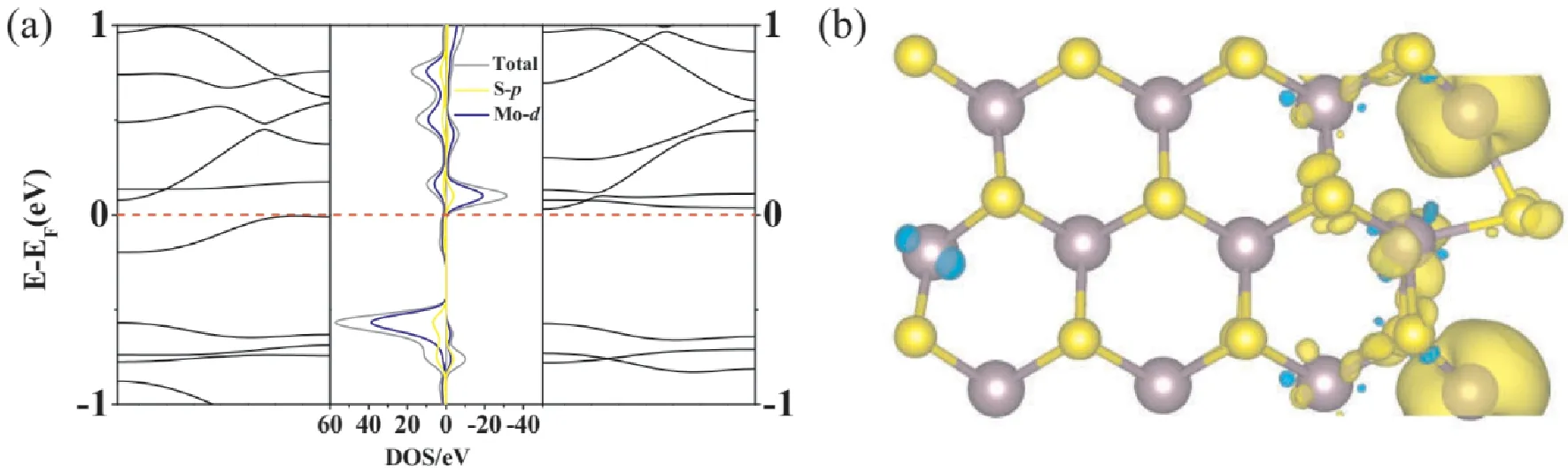
图3 (a):空位在h位点的AMoS2 NRs的能带结构和原子分波态密度图;(b):空位在h位置的自旋密度图,等值线为0.02 eV/Å3.Fig.3 (a):Energy band structure and atomic partial state density diagram of AMoS2 NRs with vacancy in h site;(b):spin density diagram of vacancy in h,contour line is 0.02 eV/Å3.
此外,我们进一步研究了空位浓度对电磁性质的影响,如图4(a),我们仍然采用带宽N=8的AMoS2NRs的结构,但将晶胞扩大两倍,并且在边缘只保留一个S空位在h位点,即S空位的浓度降低.图4(b)的能带图表明AMoS2NRs显示出半导体的性质,但其自旋向上通道和自旋向下通道并不对称,说明体系具有一定的磁性,从图4(c)自旋密度图发现磁性主要来源于S空位附近的Mo原子.
图4 (a):边缘制造了一个S缺陷的带宽N=8的双倍晶胞,(b):晶胞的能带结构和分波态密度图(PDOS),(c):晶胞的自旋密度图,等值线为0.02 eV/Å3.Fig.4 (a)Double cell with S defeat bandwidth N=8 created at the edge,(b)energy band structure and partial state density diagram(PDOS)of the cell,and(c)spin density diagram of the cell,contour line is 0.02 eV/Å3.
通过以上讨论,我们发现S空位的位置和浓度均会对AMoS2NRs电子结构和电、磁性质产生较大的影响.当S空位出现在边缘时,通过调节S空位的浓度,AMoS2NRs可以从磁性半金属调节成稀磁半导体,S空位特别是边缘的S空位改变了体系的晶体结构,从而使得边缘电荷重新分布,进而改变了其电子结构,使得体系的电、磁性质发生变化.
4 结 论
总之,第一性原理计算表明,AMoS2NRs的电子结构及自旋性质受S空位的调控.当出现S空位出现在带内部时,AMoS2NRs的物性不变,但当S空位出现在带边缘位置时,AMoS2NRs从非磁性半导体被调节成半金属;且随着S空位浓度的增加,AMoS2NRs被调节成稀磁半导体,并揭示了其物性受S空位调制的微观机理.AMoS2NRs的电子态及其自旋态受S空位调制的性质为基于MoS2纳米带的新型低维纳米电子学和自旋电子学的工程应用开发提供了更丰富的可能.
