基于HPLC指纹图谱及多指标成分定量分析的不同产地当归质量特征研究
2021-08-05于春强郭子娴王梦月李晓波
杨 燕,于春强,郭子娴,王梦月,李晓波
上海交通大学药学院,上海 200240
当归Angelicae SinensisRadix为伞形科植物Angelicae sinensis(Oliv.) Diels 的干燥根,为常用中药,具有补血活血、调经止痛、润肠通便之功效[1]。作为药食同源之品,当归也常入饮食保健[2]。当归富含有挥发油、有机酸、多糖、黄酮等化学成分[3-4],具有降压平喘、调节肠胃、抗动脉硬化、抑菌、调节机体免疫功能等药理活性[5-8]。依据目前已形成的道地产区,可将当归分为岷归(甘肃)、云归(云南)、川归(四川)、窑归(湖北)4 个大类[9]。中药材中化学成分复杂,同时药材栽培地域的的地理条件、采收时间及贮藏方法等都会对其质量造成影响[10]。因此,研究不同产地当归品质差异对于当归规范化种植、保障药材质量以及临床疗效均具有重要意义。
HPLC 指纹图谱能够有效地反映中药中的化学成分信息,宏观可量化,已成为评价和控制中药质量的常用方法[11-13]。当归药材的质量控制,除了对常规指标成分(阿魏酸,藁本内酯等)进行定量分析外,目前还进行了指纹图谱结合主成分分析、聚类分析等化学计量学研究[14-16]。然而,迄今为止,对不同商品当归的质量特征缺乏系统研究。本研究基于HPLC 指纹图谱及多成分定量分析,结合主成分分析(principal component analysis,PCA)、偏最小二乘法(partial least squares discrimination analysis,PLS-DA)、Fisher 线性判别(fisher linear discrimination analysis,FLDA)等化学计量学方法,对不同产地当归质量特征进行了分析,从化学成分角度阐明不同产地当归药材的质量特征,为当归的产地溯源识别及质量控制提供了科学依据。
1 材料与仪器
1.1 材料
27 批当归样品产于甘肃、云南、四川,均为全归,经上海交通大学王梦月副教授鉴定为伞形科植物当归Angelicae sinensis(Oliv.) Diels 的干燥根(表1)。样品存放于上海交通大学药学院李晓波教授实验室。
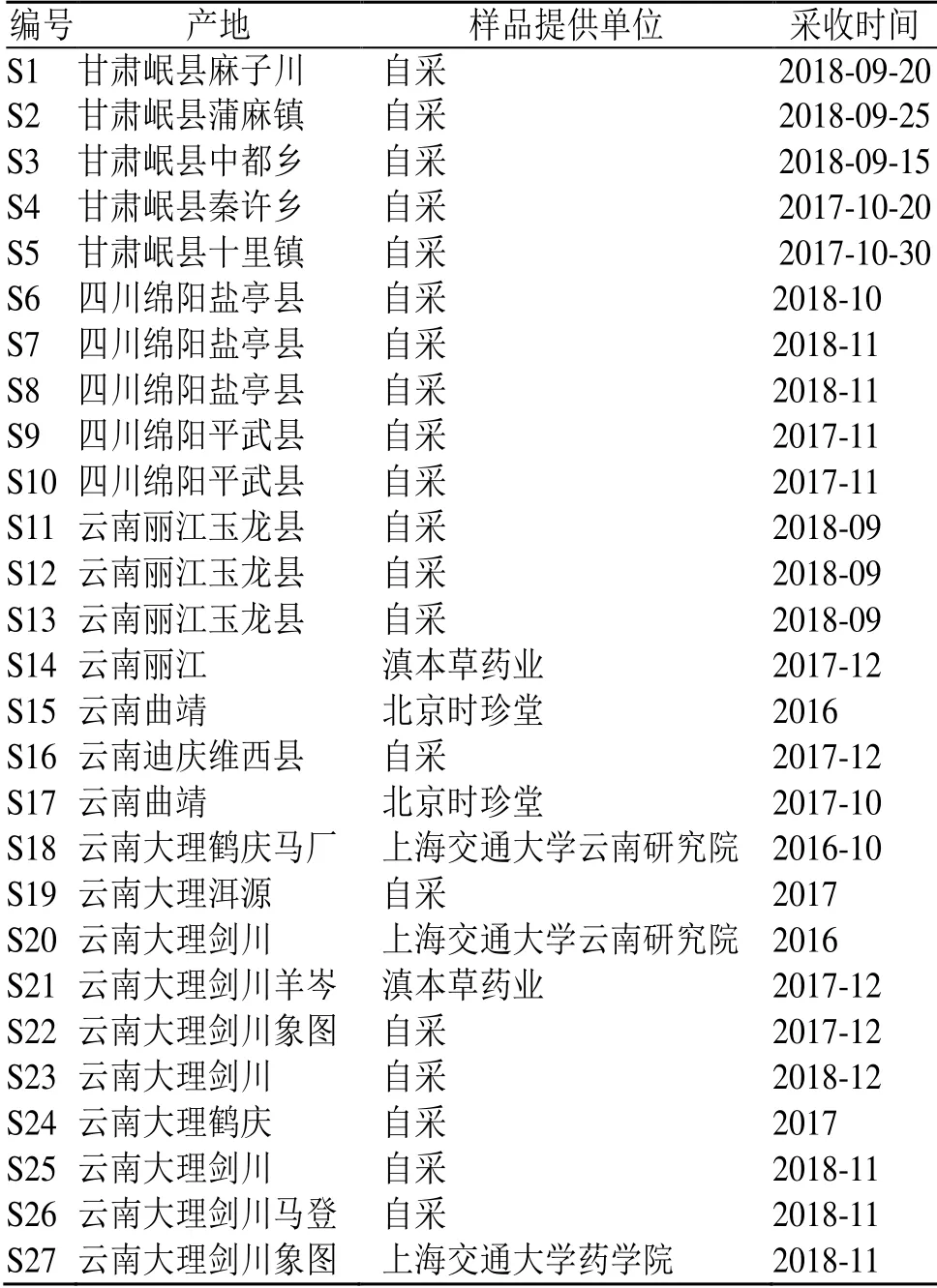
表1 不同产地27 批次当归药材样品的基本信息Table 1 Detailed information of 27 batches of Angelicae Sinensis Radix samples from different producing areas
1.2 试剂
对照品阿魏酸(批号wkq18042309)、阿魏酸松柏酯(批号 wkq18102206)、绿原酸(批号wkq18022809)、Z-藁本内酯(批号wkq18110502)、洋川芎内酯I(批号wkq18041806)、E-丁烯基苯酞(批号wkq18092104)均购至四川维克奇生物技术有限公司,质量分数均大于98%;磷酸(色谱纯,上海安谱实验科技股份有限公司);乙腈(色谱纯,北京百灵威科技有限公司);甲醇(色谱纯,北京百灵威科技有限公司);超纯水。
1.3 仪器
SB-5200 DTD 型超声波清洗仪(宁波新芝生物科技有限公司);BS-500A 型电子分析天平[赛多利斯科学仪器(北京)有限公司];BJ-500A 型粉碎机(拜杰公司);Agilent 1200 高效液相色谱仪(DAD检测器)(美国安捷伦公司);Agilent Eclipse XDB-C18 (150 mm×4.6 mm,5 μm)色谱柱(美国安捷伦公司)。
2 方法
2.1 混合对照品溶液的制备
取阿魏酸、绿原酸、阿魏酸松柏酯、洋川芎内酯I、丁烯基苯酞、Z-藁本内酯适量,精密称定,加入甲醇配置成质量浓度分别为0.666、0.724、0.770、0.696、0.664、0.536 mg/mL 的混合对照品溶液,保存于冰箱备用。
2.2 供试品溶液的制备
取当归药材约0.1 g,精密称定,置于10 mL 具塞试管中,加入5 mL 甲醇,称定质量;试管置于烧杯中,超声1 h,取出后放凉至室温,再次称定质量,用甲醇进行补足,摇匀滤过,取续滤液高效液相进样分析。
2.3 色谱条件
色谱柱为Agilent Eclipse XDB-C18(150 mm×4.6 mm,5 μm),流动相为0.1%磷酸溶液(A)-乙腈(B),体积流量1 mL/min;梯度洗脱,0~20 min,5%~30% B;20~40 min,30%~65% B;40~45 min,65% B;45~50 min,65%~100% B;50~53 min,100% B;53~58 min,100%~5% B,58~65min,5% B;检测波长254、278、316、327 nm;柱温35 ℃;进样量10 µL。
2.4 不同产地当归样品HPLC 指纹图谱研究
2.4.1 精密度试验 选取编号为S27 的当归样品约0.1 g,精密称定,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,连续进样6 次,分别计算各共有峰的保留时间与峰面积。结果表明各共有峰相对保留时间的RSD<0.85%,各共有峰相对峰面积的RSD<2.0%,表明仪器精密度良好。
2.4.2 重复性试验 选取编号为S27 的当归样品约0.1 g,精密称定,平行6 份,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,分别计算各共有峰的保留时间与峰面积,结果表明各共有峰的相对保留时间的RSD<1.0%,各共有峰的相对峰面积的RSD<1.2%。表明本方法重复性良好。
2.4.3 稳定性试验 选取编号为S27 的当归样品约0.1 g,精密称定,按“2.2”项下条件制备供试品溶液,分别于0、2、4、8、16、24 h 进样,测定方法条件同“2.3”项下,计算各共有峰的保留时间及峰面积。结果各共有峰的相对保留时间的RSD<1.8%,各共有峰的相对峰面积的RSD<2.5%。表明供试品在24 h 内稳定。
2.4.4 指纹图谱的建立及化学计量学分析 分别精密称取27 批当归药材粉末,按“2.2”项下条件制备成全归供试品溶液,测定方法条件同2.3 项下。将所得的样品数据全谱导入国家药典委员会组织定型的《中药色谱指纹图谱相似度评价系统(2012版)》软件中,采用中位数法,时间窗口设定为0.5 min,选取S1 作为参照图谱(R),生成当归药材中药指纹图谱,并进行相似度计算。将得到的13 个共有峰的峰面积导入SPSS(23.0 版本)、SIMCA-P(13.0 版本)进行PCA 统计分析、逐步线性判别,使用SIMCA-P 进行有监督模式的PLS-DA 分析,进行自动拟合求解。使用Prism GraphPad 8.0 作图。
2.5 指标成分含量测定
2.5.1 方法学考察
(1)线性关系:将“2.1”项下配制的混合对照品溶液进行梯度稀释,配制为6 份不同浓度的混合对照品溶液。按照“2.3”项下条件进行测定,以对照品质量浓度为横坐标(X),以各对照品峰面积为纵坐标(Y)绘制标准曲线,计算回归方程及相关系数(r),结果见表2,各成分在浓度范围内线性关系良好。
(2)最低检测限(LOD):将对照品溶液稀释多份,按照“2.3”项下条件进行测定,以信噪比(S/N)为3∶1 时的溶液浓度作为LOD,结果见表2。
(3)最低定量限(LOQ):将对照品溶液稀释多份,按照“2.3”项下条件进行测定,以信噪比(S/N)为10∶1 时的溶液浓度作为LOQ,结果见表2。

表2 线性关系、LOD 和LOQ 考察结果Table 2 Detailed information of standard curve, LOD and LOQ
(4)精密度试验:选取编号S27 的当归样品约0.1 g,精密称定,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,连续进样6 次,分别计算各共有峰的保留时间与峰面积。结果表明阿魏酸、阿魏酸松柏酯、绿原酸、Z-藁本内酯、洋川芎内酯I、E-丁烯基苯酞RSD 依次为0.67%、0.63%、0.43%、0.98%、1.20%,0.48%,表明仪器精密度良好。
(5)重复性试验:选取编号S27 的当归样品约0.1 g,精密称定,平行6 份,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,分别计算各共有峰的保留时间与峰面积,结果表明各共有峰的相对保留时间的RSD<1.0%,各共有峰的相对峰面积的RSD<1.2%。表明本方法重复性良好。
(6)稳定性试验:选取编号为S27 的当归样品约0.1 g,精密称定,按“2.2”项下条件制备供试品溶液,分别于0、2、4、8、12 、24 h 进样,测定方法条件同“2.3”项下,计算各共有峰的保留时间及峰面积。结果各共有峰的相对保留时间的RSD<1.8%,各共有峰的相对峰面积的RSD<2.5%。表明供试品在24 h 内稳定。
(7)加样回收率试验:取S27 号当归样品约0.1 g,精密称定,共计6 份,每份都大致以1∶1 的比例加入相应化学成分的对照品,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,分别记录各共有峰的保留时间与峰面积,计算各成分的加样回收率和RSD 值。阿魏酸、阿魏酸松柏酯、绿原酸、Z-藁本内酯、洋川芎内酯I、E-丁烯基苯酞的加样回收率分别为95.5%、93.2%、95.4%、95.0%、91.1%、93.2%;RSD 值分别为1.28%、1.17%、2.01%、0.77%、3.37%、2.18%。
2.5.2 样品测定:取当归样品约0.1 g,精密称定,按“2.2”项下条件制备供试品溶液,测定方法条件同“2.3”项下,记录峰面积,按标准曲线计算各待测成分的含量。
3 结果与分析
3.1 HPLC 指纹图谱的建立
将27 批次样品色谱数据导入“中药色谱指纹图谱相似度评价系统”,建立27 批样品的HPLC 指纹图谱(图1),以S1 为参照图谱,时间窗口设定为0.5 min,以中位数法建立对照图谱(图2),共得到稳定性较好的13 个色谱峰作为共有峰。取当归对照品溶液,按照“2.3”项下分析条件进样测定,通过与混合对照品溶液色谱图比对、查阅文献资料共指认出8 个色谱峰,其中2 号峰为绿原酸,3 号峰为阿魏酸,4 号峰为洋川芎内酯I,5 号峰为洋川芎内酯H,9 号峰为阿魏酸松柏酯,10 号峰为E-藁本内 酯,11 号峰为E-丁烯基苯酞,12 号峰为Z-藁本内酯。藁本内酯的出峰时间稳定且峰面积占比较大,作为参比峰,计算各共有峰的相对保留时间及相对峰面积。
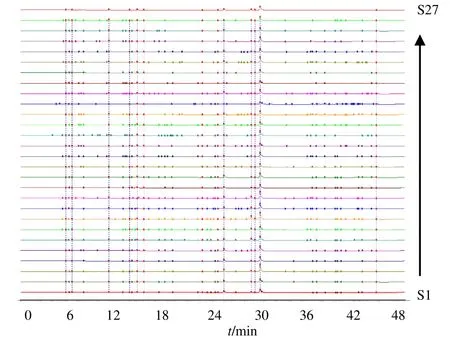
图1 27 批次当归HPLC 指纹图谱Fig.1 HPLC fingerprint of 27 batches of Angelicae Sinensis Radix
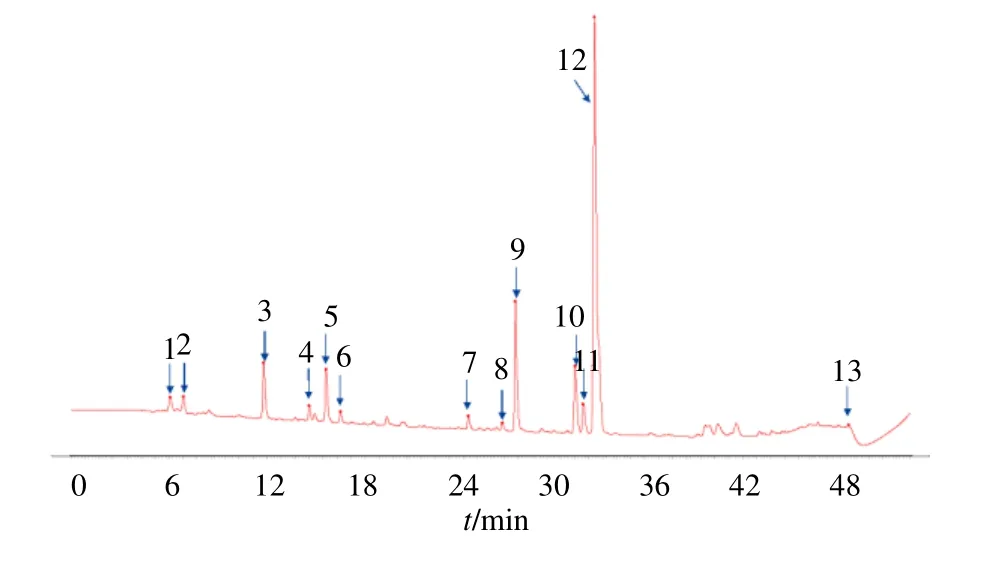
图2 当归药材HPLC 对照指纹图谱(中位数法)Fig.2 Reference HPLC fingerprint of Angelicae Sinensis Radix (median method)
3.2 不同产地当归的化学模式识别
3.2.1 相似度评价 采用《中药色谱特征图谱相似度评价系统软件(2012 版)》对27 批次的当归药材色谱数据进行分析,生成指纹图谱(图1),并计算得到相似度评价结果,结果表明27 批次当归药材各色谱图相似度在0.900~0.998。各批次药材与参照图谱S1 比较时,S6、S7、S15、S18、S20、S21、S24、S26 相似度较为离散,表明各个产地当归之间存在一定差异。
3.2.2 PCA 通过主成分分析共提取了4 个特征值大于1 的主成分,由当归药材指纹图谱共有峰特征值(表4)可知,4 个主成分的累积方差贡献率达到85.260%,说明4 个主成分在反映3 个产地的当归样品共有成分的关系中起到主导作用。其中第1 个主成分贡献率为34.751%;第2 个主成分贡献率为31.718%;第3、4 个主成分贡献率分别为10.595%、8.197%。由主成分矩阵(表5)可知各个共有峰对4 个主成分不同的贡献率,第1 主成分主要代表了峰2~4、10~12;第2 主成分主要代表了峰4~8、13;第3 主成分的信息主要来自峰9、12、13;第4 主成分的信息主要来自峰1。同时采用多元统计分析软件 SIMCA-P 14.0 对13 个共有峰进行PCA 分析(图3),可见样品被分为3 类:第1 类为S1~S5,第2 类为S6~S10,第3 类为S11~S27,此分类结果与按当归产地分类结果一致,表明前4 个主成分可以代表27 批次样品的整体质量。

表4 当归药材指纹图谱共有峰特征值Table 4 Characteristic values of common peaks in HPLC fingerprint of Angelicae Sinensis Radix

表5 共有峰成分矩阵Table 5 Component matrix of common peaks
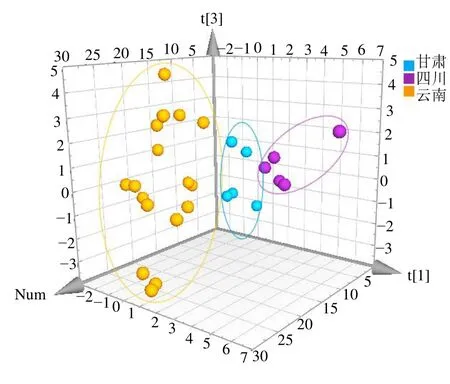
图3 27 批次当归主成分分析Fig.3 Principal component analysis of 27 batches of Angelicae Sinensis Raidx
3.2.3 PLS-DA 由PLS-DA 三维得分图(图4)可知,3 个产区当归可明显区分,与PCA 结果一致,且组别间分离效果更加显著,进一步通过计算各个共有峰的变量重要性投影(Variable importance in the projection,VIP)值(图5),以VIP 值大于1 为标准,筛选可以作为产地间相互区分的差异性指标,结果显示Z-藁本内酯(VIP=2.08),E-藁本内酯(VIP=1.83),阿魏酸松柏酯(VIP=1.75),绿原酸(VIP=1.00)4个成分是可以作为组间区分的潜在指标,与主成分分析结果也较为接近。
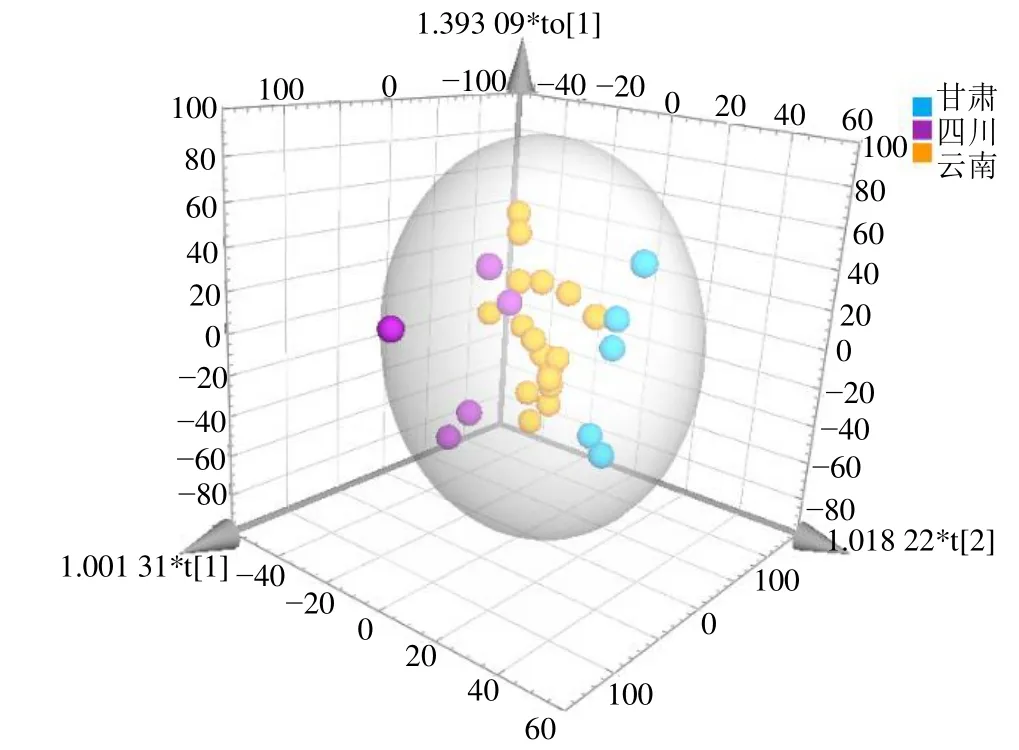
图4 27 批次当归PLS-DA 得分图Fig.4 PLS-DA scores of 27 batches of Angelicae Sinensis Radix
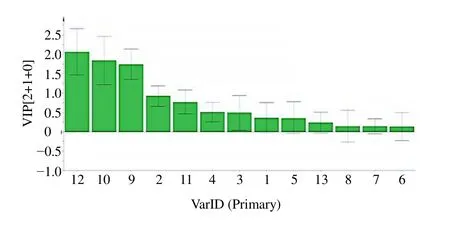
图5 27 批次当归13 个共有峰VIP 值Fig.5 VIP values of 13 common peaks of 27 batches of Angelicae Sinensis Radix
3.3 不同产地当归的化学成分含量测定及判别分析验证
3.3.1 当归中6 个指标成分含量测定 由当归HPLC 指纹图谱共指认出13 个共有化学成分,存在VIP 值大于1 的4 个差异性成分,其中藁本内酯中Z型相比E型为优势构象,比E型更加稳定,在中药材中含量也相对较高;阿魏酸松柏酯由于不稳定易转化为阿魏酸与松柏醇,在实验中将阿魏酸松柏
酯含量换算为等量阿魏酸,并与本身测定的阿魏酸之和计入总量,以“总阿魏酸”表示。因此本试验主要测定了当归样品中总阿魏酸、Z-藁本内酯、绿原酸的含量,同时考虑到当归中的洋川芎内酯I,E-丁烯基苯酞也是主要有效成分,因此一并纳入进行含量测定,结果见表6,可见不同产地当归中化学成分含量存在明显差异。通过分析含量测定结果箱线图(图6),可明确各产地当归中代表性化学成分。四川当归中的绿原酸量均在0.30 mg/g 以上,整体高于其他产地(P<0.05),可作为区分四川当归与其他产地当归的一个指标成分;洋川芎内酯I在云南迪庆维西县采收的当归中质量分数最高,达到0.24 mg/g;四川产区的当归中E-丁烯基苯酞量基本都在0.50 mg/g 以上,高于其他产地当归(P<0.000 1),也可作为区分四川当归与其他产地当归的一个指标成分,甘肃岷县当归中含量在0.2~0.3 mg/g,组内差异不明显,云南产区的S16、S18、S19、S20、S21、S24 号当归E-丁烯基苯酞含量高于云南产地的其他批次当归;27 批次当归中总阿魏酸的质量分数均在1.42~2.58 mg/g,其中S8、S23、S25、S26、S27 5 个批次中总阿魏酸的含量均达到了2.50 mg/g 以上,剑川当归中阿魏酸含量明显高于其他产区(P<0.05),可作为区分云南剑川当归与其他产地当归的差异性成分;7 个批次的云南剑川当归中Z-藁本内酯基本都在23.00 mg/g 以上,云南大理洱源批次当归则为最低,甘肃岷县麻于乡的当归中质量分数最高,达到29.15 mg/g,岷县蒲麻镇与中都乡产地当归的质量分数也均在27.00 mg/g以上,高于其他产地(P<0.05),可作为区分甘肃当归与其他产地当归的差异性成分。

表6 当归中指标成分测定结果Table 6 Determination results of marker components in Angelicae Sinensis Radix

图6 HPLC 指纹图谱各指标成分含量测定箱线图Fig.6 Determination results of marker components of HPLC fingerprint from Angelicae Sinensis Radix
3.3.2 FLDA 利用SPSS 23.0 统计软件的Fisher 线性判别分析验证指标成分可以作为当归产地识别的适用性,对来自甘肃、四川以及云南的27 个样本按地区进行分组建模,以绿原酸、洋川芎内酯I、E-丁烯基苯酞、总阿魏酸、Z-藁本内酯作为判别变量,根据Wilks’Lambda 统计量,当被加入的变量F值>3.84 时,该变量则可以保留在函数中建立判别函数,最终提取到绿原酸(a)、E-丁烯基苯酞(b)、Z-藁本内酯(c)3 个变量,建立2 个判别函数(discriminant function,DF)。

DF(1)携带原始信息的 83.6%,DF(2)携带原始信息的16.4%。以2 个判别函数得分作图,获得典则质心图(图7),可以看出各产地样 本之间各自聚为一类,且存在组间距离,3 个成分对当归产地溯源具备区分潜力。由样本自身验证及交叉验证结果,自身验证判别中只有甘肃产地中一个样本被误判为云南产地,样本总正确判别率达96.3%;样本交叉验证结果中甘肃产地中一个样本被误判为云南产地,云南产地中一个样本被误判为四川当归样本,样本总正确判别率达92.6%。表明绿原酸、Z-藁本内酯、E-丁烯基苯酞可以作为产地区分的指标成分,这一结果与PLS-DA 分析、及含量测定分析结果基本一致。

图7 判别分析典则质心图Fig.7 Centroid of canonical discriminant function
4 讨论
本实验利用二极管阵列检测器对当归样品供试液进行全波长扫描,获得3D 图谱,查看色谱峰的紫外吸收光谱,结果表明当检测波长设定为254 nm时,色谱基线平稳,色谱峰分离度良好,色谱峰数目多。通过设计正交试验,考察超声提取条件。以阿魏酸、阿魏酸松柏酯、绿原酸、E-丁烯基苯酞、洋川芎内酯I、Z-藁本内酯的峰面积为评价指标,对原始数据均值化,然后取6 个指标的平均值作为每次试验的试验结果。结果显示,50 倍的100%甲醇超声60 min 为最优提取条件。
当归品质的形成与非生物因素息息相关,如水分、光照、海拔等。据考证,当归最早的出产地在今甘肃宕昌一带,是公认的当归道地产区[17],随着栽培技术的发展,云南、四川等地的当归产量也逐步提升,但单纯从当归的外观性状、HPLC 指纹图谱中很难明确其产地来源及内在质量差异。《中国药典》2015年版中将当归药材的阿魏酸含量作为质量控制指标[1]。但是在当归中的阿魏酸有2 种来源,一是植物本身含有的阿魏酸,即游离阿魏酸,另一种是由其他化合物经过某些途径转化为阿魏酸,阿魏酸松柏酯性质很不稳定,在甲醇、强酸性、强碱性、光照及高温等环境下易转化为阿魏酸与松柏醇[18],药典中测定的阿魏酸含量会高于当归本身含有的阿魏酸,它实际上包括了游离阿魏酸以及由阿魏酸松柏酯转化而来的阿魏酸的总量;同时在考察当归样品提取的提取工艺时,若采用回流提取,阿魏酸松柏酯会由于受热不稳定分解,导致样品重现性较差;采用超声提取,可减少阿魏酸松柏酯由于加热造成分解挥发,并将阿魏酸及阿魏酸松柏酯以总阿魏酸的量计入,可避免由于环境条件改变造成阿魏酸含量发生改变,含量测定不准确的情况。此外,当归中还存在其他活性成分,如苯酞类的藁本内酯、丁烯基苯酞,有机酸类的阿魏酸、绿原酸等成分均表现出较强的药理活性作用[11-13],仅测定阿魏酸的含量,当归质量特征得不到全面的表征与反映。
本研究结果表明,云南产区当归中总阿魏酸的含量整体高于其他产区,特别是剑川当归中总阿魏酸的含量均在2.00 mg/g 以上,可作为云南剑川当归区别于其他产区的特异性指标成分,相似结论也有研究报道[19-20]。当归中的挥发性成分种类繁多,主要包括藁本内酯、正丁烯基内酯等,现代医学认为,当归挥发性成分在解痉、阵痛、抗炎、抑制血小板聚集等方面都有药理活性[5]。甘肃产区当归中挥发性成分Z-藁本内酯可作为甘肃当归的成分识别标志;四川当归中除了E-丁烯基苯酞之外、绿原酸含量也明显高于其他产区,可作为四川当归的成分识别标志。本实验注重于不同产区当归内在质量差异,由PCA、PLS-DA 寻找到的潜在性差异性成分可实现不同产地当归的区分溯源,并对各个产地当归的特异性识别成分进行归属,并经由判别分析验证结果可靠性,寻找到不同产地当归的“识别标志”,以期能为当归药材的质量控制及产地识别提供依据,对于当归规范化种植、提升药材质量和临床疗效上也有积极意义。
利益冲突所有作者均声明不存在利益冲突
