超高效液相色谱-串联质谱法测定酱卤肉制品中1-甲基咪唑、2-甲基咪唑及4-甲基咪唑
2021-06-18闵宇航黄璐瑶余晓琴
闵宇航,黄璐瑶,余晓琴,王 颖
(四川省食品药品检验检测院,四川成都 610097)
焦糖色是一种食品添加剂,不仅能起到增色增香的作用,还有助于提高食品品质,被广泛应用于酱油、食醋、料酒、烘焙食品、糖果、饮料等多种食品的生产中。焦糖色按生产工艺可以分为四类:Ⅰ类为普通法焦糖色,Ⅱ类为苛性亚硫酸盐法焦糖色,Ⅲ类为氨法焦糖色,Ⅳ类为亚硫酸铵法焦糖色。在焦糖色的生产过程中主要发生了美拉德反应和焦糖化反应。普通的焦糖色主要发生焦糖化反应,而加入了氨(铵)作为催化剂的Ⅲ、Ⅳ类焦糖色以美拉德反应为主。与非氨(铵)法生产相比,氨(铵)法生产焦糖色具有反应快、周期短、呈色深的特点,但是会产生美拉德反应的伴生危害产物2-甲基咪唑和4-甲基咪唑。
4-甲基咪唑具有较强的惊厥作用,可以使动物产生超兴奋状态从而发生痉挛[1]。2007年美国国家毒理学规划处(National Toxicology Program,NTP)将4-甲基咪唑作为可致癌物质发出警告[2],美国加州环境健康危害评估办公室(OEHHA)2011年将4-甲基咪唑列入致癌化学品清单(the Proposition 65 list)中,并设定4-甲基咪唑的致癌潜能为0.024 (mg/kg bwday),无显著风险水平(NSRL)为29 μg/d,要求超过该安全值的食品必须带有警告性标识[3]。2013年,世界癌症研究机构(IARC)致癌物分级目录将4-甲基咪唑、2-甲基咪唑列入2B级致癌物[4]。1-甲基咪唑与4-甲基咪唑、2-甲基咪唑互为同分异构体,其毒理学性质没有得到充分的调查,但在部分食品中发现1-甲基咪唑的存在[5]。酱卤肉制品是日常消费量较大的食品,但是目前并没有酱卤肉制品中甲基咪唑类化合物含量的研究报道。GB 2760-2014中规定4类焦糖色均不能在酱卤肉制品中使用[6],酱卤肉制品中的甲基咪唑类污染物可能来源于生产者非法添加了Ⅲ、Ⅳ类焦糖色,也可能来源于配料中酱油、醋等调味品的带入。
目前报道食品中检测的甲基咪唑类化合物多为2-甲基咪唑和4-甲基咪唑,缺少1-甲基咪唑的检测。检测方法有分光光度法[7-8]、拉曼光谱法[9]、气相色谱法[10-12]、气相色谱质谱联用法[13-15]、高效液相色谱法[16-17]和液相色谱质谱联用法[18-20]。分光光度法和气相色谱法的前处理较为繁琐[21-22];拉曼光谱仪普及度较低;高效液相色谱法的灵敏度较低。本实验期望结合超高效液相色谱的快速分离能力以及质谱的高选择性、高灵敏度,建立一种快速、准确、稳定的UPLC-MS/MS法测定酱卤肉制品中的1-甲基咪唑、2甲基咪唑和4-甲基咪唑,填补酱卤肉制品中甲基咪唑类化合物检测的空白,为以后监管部门的风险评估和限值制定打下了基础。
1 材料与方法
1.1 材料与仪器
Waters Oasis MCX固相萃取小柱(150 mg,6 mL) 美国Waters公司;1290 Infinity Ⅱ超高效液相色谱仪、 6460C三重四极杆质谱仪 美国Agilent公司;Thermo Scientific HeraeusMlutifuge X3R高速冷冻离心机 美国Thermo Fisher Scientific公司;IRM IDH30超声仪 德国IRM Technology GmbH公司;IKA MS3涡旋混合器 德国IKA公司;BiotageTurboVap全自动氮吹仪 瑞典Biotage公司;HM100 POWTEQ刀式研磨仪 北京格瑞德曼公司。
1.2 实验方法
1.2.1 标准溶液的制备 准确称取1-MEI、4-MEI标准品10 mg(精确至0.01 mg),用乙腈溶解并定容至10 mL,混匀,制成1 mg/mL的标准储备液。精密吸取10 μL 1-MEI、4-MEI标准储备液以及100 μL 2-MEI的标准品置10 mL容量瓶中,乙腈定容至刻度,混匀,制成1 μg/mL的混合标准溶液。
准确称取1-MEI-D6、2-MEI-D6、4-MEI- D6标准品10 mg,用乙腈溶解并定容至10 mL,混匀,制成1 mg/mL的内标储备液。再精密吸取10 μL 1-MEI-D6、2-MEI- D6、4-MEI- D6的内标储备液置10 mL容量瓶中,乙腈定容至刻度,混匀,制成1 μg/mL的混合内标溶液。
精密吸取混合标准溶液和混标内标溶液适量,用初始流动相稀释成30、60、100、150、200、300、400 ng/mL混合系列标准工作溶液,其中1-MEI-D6、2-MEI- D6、4-MEI- D6的浓度均为50 ng/mL。
1.2.2 供试品溶液的制备 取样品约200 g,用刀式研磨仪充分均质,装入洁净的容器中。
称取上述均质后的样品2 g(精确至0.001 g),置50 mL离心管中,加入混合内标溶液100 μL,加水20 mL,涡旋1 min,再超声(功率500 W)5 min,以10000 r/min在4 ℃条件下离心5 min,上清液待净化。
净化前,依次用5 mL甲醇,5 mL水对MCX固相萃取小柱进行活化。再精密量取10 mL上清液,过MCX固相萃取小柱,依次用2%甲酸水溶液5 mL,甲醇5 mL淋洗,再用5%氨水甲醇溶液10 mL洗脱,收集全部洗脱液,45 ℃氮吹至近干,精密加入初始流动相1 mL复溶,过0.22 μm有机系滤膜,滤液供液相色谱-串联质谱仪测定。
1.2.3 分析条件 液相色谱条件:色谱柱:Agilent Zorbax HILIC色谱柱(2.1 mm×100 mm,3.5 μm);柱温:35 ℃;进样量:5 μL;流动相:A相:乙腈;B相:5 mmol/L乙酸铵溶液;流速0.6 mL/min;流动相梯度见表1。
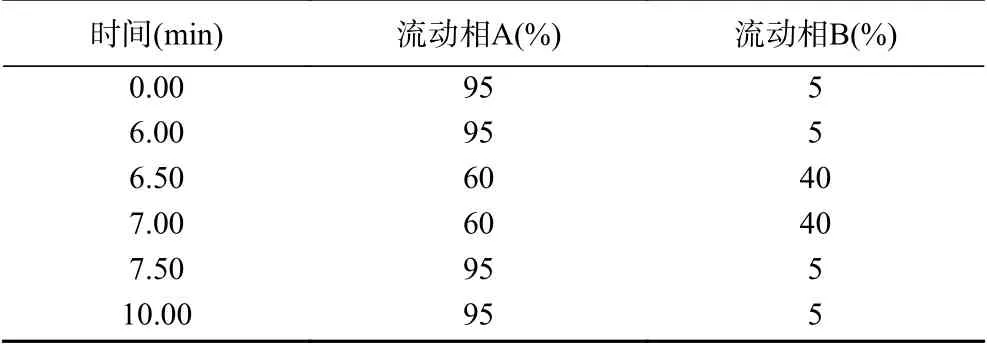
表1 流动相梯度洗脱表Table 1 Gradient elution program
质谱条件:离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;检测方式:多反应检测(MRM);毛细管电压:3.5 kV;干燥气温度:325 ℃;干燥气流量:11 L/min;雾化气压力:35 psi;鞘气温度:325 ℃;鞘气流量:11 L/min。化合物质谱参数见表2。
“知识改变命运”,这是现代人公认的真理。可惜的是,在懵懂的转型时期,经受着阵痛的人们对此并没有清晰的认知。在此情况下,他们既无法与伴侣进行良好的沟通,更难以摆脱贫困潦倒的生存现实。因为不明就里,即便有偶而的挣脱,也显得苍白无力。
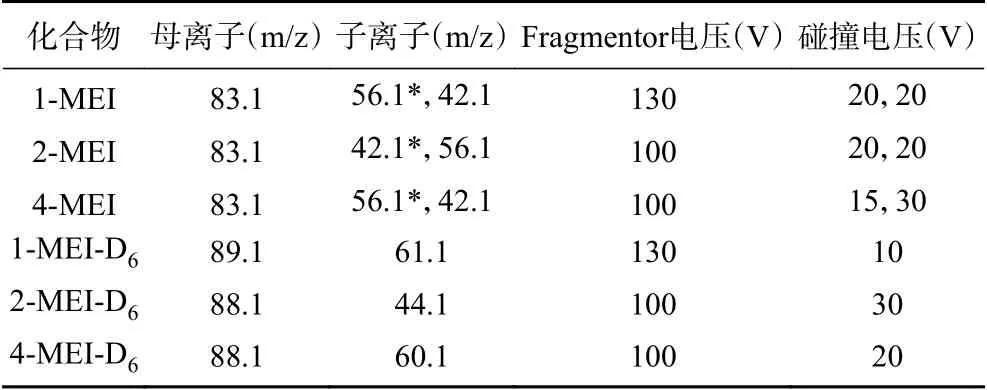
表2 1-MEI、2-MEI、4-MEI、1-MEI-D6、2-MEI-D6、4-MEI-D6的质谱参数Table 2 Mass parameters of 1-MEI, 2-MEI, 4-MEI,1-MEI-D6, 2-MEI-D6, 4-MEI-D6
1.3 数据处理
采用美国Agilent Technologies Inc.的MassHunter Workstation Software 06.00软件进行分析处理,Microsoft Office Excel软件进行统计分析。
2 结果与分析
2.1 色谱条件的优化
2.1.1 色谱柱的选择 1-MEI、2-MEI、4-MEI这3种化合物互为同分异构体,具有相同的分子质量和离子碎片,需要通过色谱柱实现完全分离后才能进行检测[23]。由于目标化合物为含氮极性小分子化合物,采用常用的十八烷基硅烷键合硅胶柱可能会出现保留差、出峰快、分离效果不佳的情况[24]。实验首先尝试了Merck Purospher STAR LP RP-18 endcapped(4.6 mm×250 mm,5 μm),3种化合物在填料为C18的色谱柱上无法达到很好的分离效果。实验又选择了适合高极性化合物的HILIC填料的色谱柱进行分离。分别比较了Thermo Acclaim Mix-Mode HILIC-1(2.1 mm×150 mm,3 μm)、Agilent Zorbax HILIC Plus(2.1 mm×100 mm,3.5 μm)、Agilent Poroshell 120 HILIC(2.1mm×100 mm,2.7 μm)、Waters ACQUITY UPLC BEH HILIC(2.1 mm×100 mm,1.7 μm)4种不同品牌和参数的HILIC色谱柱。最终选择了Agilent Zorbax HILIC(2.1 mm×100 mm,3.5 μm),可以实现目标化合物的很好分离(见图1)。
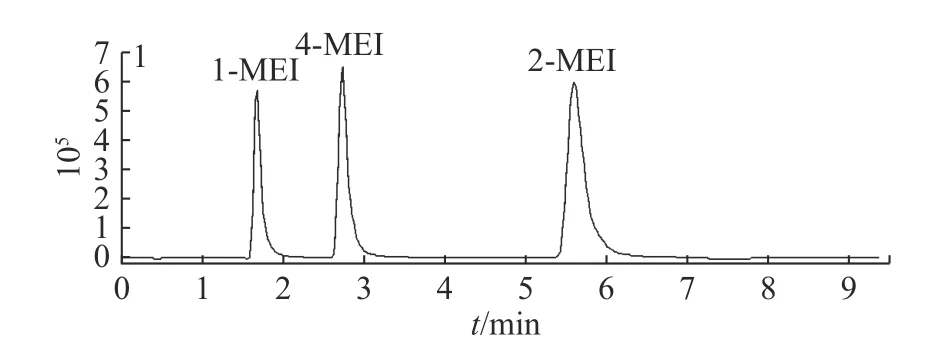
图1 酱卤肉基质中1-MEI, 2-MEI和4-MEI的色谱图Fig.1 Chromatogram of 1-MEI, 2-MEI and 4-MEI in sauced meat products
2.1.2 流动相的优化 乙腈是HILIC色谱柱常用的有机流动相,而在流动相水相中加入缓冲盐有利于提高色谱柱的柱效和重复性[25-26]。实验比较了不同梯度条件的乙腈-5 mmol/L的乙酸铵溶液。在表1的梯度条件下,目标化合物可以达到一个很好的分离。
2.2 质谱条件的优化
分别取用1-MEI、2-MEI、4-MEI、1-MEI-D6、2-MEI-D6、4-MEI-D6标准溶液,直接进样进行质谱条件优化。在ESI +模式下分别进行全扫描,确定1-MEI、2-MEI、4-MEI的母离子均为m/z 83.1,对其母离子进行二级质谱子离子扫描,3种待测化合物均分别丢失-C2H3和-NC2H3,相应产生碎片离子m/z 56.1和m/z 42.1;在ESI+模式下分别进行全扫描,1-MEI-D6、2-MEI-D6、4-MEI-D6均出现m/z 89.1和m/z 88.1两个母离子峰,1-MEI-D6的m/z 89.1响应高于m/z 88.1,实验选择m/z 89.1作为1-MEID6的母离子;而2-MEI-D6和4-MEI-D6的m/z 88.1响应高于m/z 89.1,实验选择m/z 88.1作为2-MEI-D6和4-MEI-D6的母离子。对三种内标化合物的母离子进行二级质谱子离子扫描,分别产生碎片离子m/z 61.1、m/z 60.1、m/z 44.1,作为内标化合物的定量离子。
2.3 前处理条件的优化
2.3.1 提取溶剂的选择 根据3种甲基咪唑类化合物物理和化学性质,实验采用添加水平为50 μg/kg的基质加标实验,每个考察条件做6个平行实验(n=6)。考察水、甲醇、乙腈3种提取溶剂的提取能力,以加标回收率考察提取效果(见图2)。结果表明,1-MEI:水>乙腈>甲醇;4-MEI:水>甲醇>乙腈;2-MEI:水与甲醇效果相似,均明显优于乙腈。综上,同时考虑成本、环保等因素,选择水为提取溶剂。
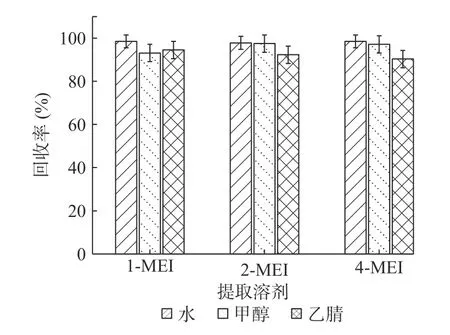
图2 提取溶剂的考察(n=6)Fig.2 Research of extraction solvents(n=6)
2.3.2 固相萃取小柱的选择 甲基咪唑类化合物由于其分子中含有氮原子,净化时宜采用对碱性物质有较好保留的阳离子交换固相萃取柱[27]。因此实验比较了市面上常见的两种品牌的阳离子交换固相萃取小柱,分别为Waters Oasis MCX(150 mg,6 mL)和Agela Cleanert PCX(150 mg,6 mL)。采用添加水平为50 μg/kg的基质加标实验(n=6),以加标回收率考察两个小柱的净化效果(见图3)。结果表明Waters Oasis MCX净化效果略优于Agela Cleanert PCX。
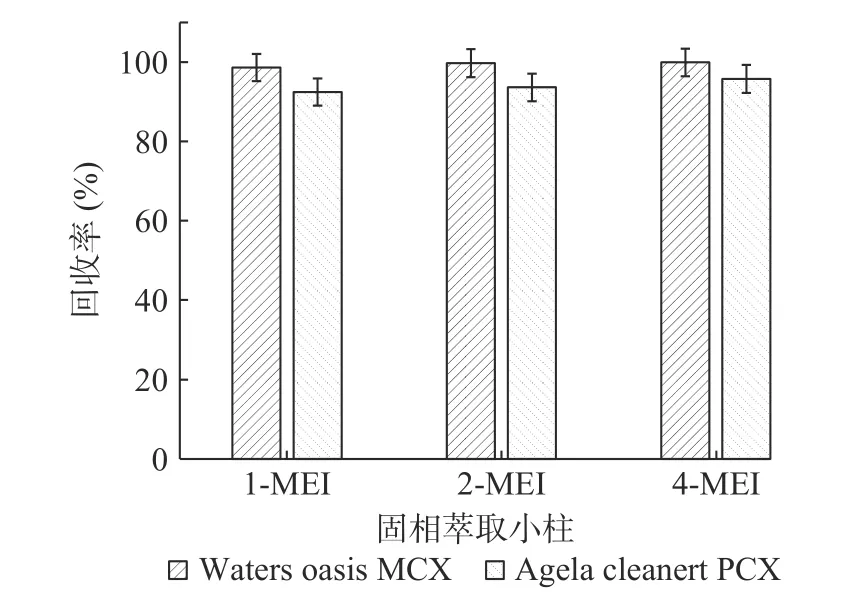
图3 固相萃取小柱的考察(n=6)Fig.3 Research of solid phase extraction colum(n=6)
2.3.3 固相萃取条件的优化 实验采用添加水平为50 μg/kg的基质加标实验(n=6),以加标回收率考察了不同浓度的氨水-甲醇溶液对目标化合物的洗脱能力,分别考察了2%、5%、10%、20%的氨水-甲醇溶液(见图4)。四种溶液对2-MEI、4-MEI的洗脱效果无明显差异,其中1-MEI洗脱受氨水浓度的影响稍大,从实验结果来看,选择5%氨水-甲醇溶液作为洗脱溶剂。
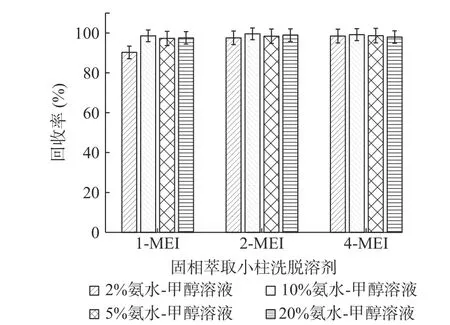
图4 MCX固相萃取小柱洗脱溶剂的考察(n=6)Fig.4 Research of the elution solvent on MCX solid phase extraction column(n=6)
实验考察了不同体积的洗脱溶剂的对目标化合物的洗脱能力。分别考察了5、10、15 mL的洗脱溶剂用量,以加标回收率考察不同用量洗脱溶剂的洗脱效果(见图5)。结果发现使用10 mL及15 mL洗脱溶剂的洗脱效果接近,均优于5 mL的洗脱效果。综合洗脱效果、成本及环保等因素,选择10 mL作为洗脱体积。
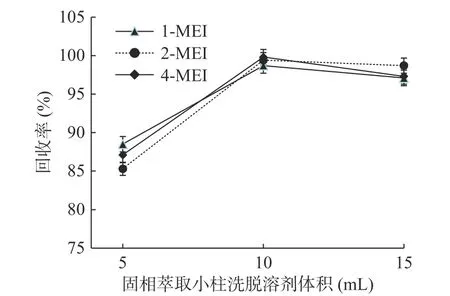
图5 MCX固相萃取小柱洗脱溶剂体积的考察(n=6)Fig.5 Research of the volume of the elution solvent(n=6)
2.4 方法学验证
2.4.1 基质效应 实验通过提取后添加法来评价基质效应(matrix effect, ME),将空白样品按前处理方法进行提取净化后,在空白样品提取液中添加目标化合物,用已定的色谱、质谱条件进行检测,然后与同样浓度的纯溶剂中目标化合物的离子响应强度进行比较。采用公式:其中A和B分别表示纯溶剂与基质溶液中分析物的离子响应强度。若ME<1,说明基质对分析物的响应产生了抑制作用;ME>1,说明基质会增强分析物的响应;ME=1,说明不存在基质效应。实验考察了三个浓度水平的基质效应(30、200、400 ng/mL),1-MEI三个浓度水平下的ME分别为0.65、0.55、0.57;2-MEI三个浓度水平下的的ME分别为0.78、0.74、0.77;4-MEI三个浓度水平下的的ME分别为0.54、0.57、0.58.结果说明基质对3种咪唑类化合物均有程度不一的抑制作用。实验采用同位素内标法定量以消除基质效应的影响,保证定量的准确性[28-29]。
2.4.2 线性范围、检出限和定量限 用初始流动相配制1-MEI、2-MEI和4-MEI的混合系列标准工作溶液,浓度分别为30、60、100、150、200、300、400 ng/mL。以目标化合物定量离子的峰面积与相应内标化合物定量离子的峰面积的比值为纵坐标,质量浓度为横坐标得到线性回归方程(见表3),3种甲基咪唑化合物线性相关系数均大于0.9997。通过空白样品加标测定,3种甲基咪唑类化合物在10 μg/kg的添加水平下经过前处理所得试液的定性离子信噪比(S/N)均大于3,表明该方法的3种甲基咪唑类化合物的检出限均可达到10 μg/kg;3种甲基咪唑类化合物在30 μg/kg的添加水平下经过前处理所得试液的定性离子信噪比(S/N)均大于10,且1-MEI、2-MEI和4-MEI的回收率分别为106.0%、100.6%、92.0%,表明该方法3种化合物的定量限均可达到30 μg/kg。
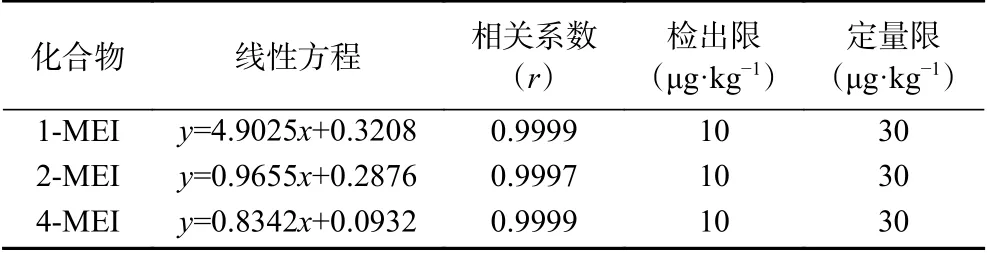
表3 1-MEI、2-MEI和4-MEI的线性方程、相关系数、检出限及定量限Table 3 Regression equation, correlation coefficient, limits of detection (LOD) and limits of quantitation(LOQ) of 1-MEI, 2-MEI and 4-MEI
2.4.3 回收率、精密度和稳定性 选取酱卤肉样品中添加30、60、300 μg/kg三个水平,每个水平分别做6份平行样,计算回收率和相对标准偏差(RSD)。3种甲基咪唑类化合物三个加标水平下的平均回收率为92.0%~116.3%,RSD为0.70%~3.96%。样品加标溶液分别在0、2、4、8、16、24 h进样,3种甲基咪唑类化合物的定量离子峰面积RSD为2.69%~4.41%。结果表明该方法的准确、稳定、可靠。
2.4.4 实验室间协作验证 为验证本方法的有效性和适用性,选择了6家检测机构对本方法进行验证。6家检测机构的结果显示,3种甲基咪唑类化合物在30~400 ng/mL范围内线性关系良好,相关系数(r)均大于0.9990;方法检出限浓度下的定性离子信噪比均大于3;方法定量限浓度下的定性离子信噪比均大于10。30、60、300 μg/kg三个加标水平的平均回收率为77.2%~115.2%,RSD为0.80%~9.34%。
2.5 实际样品的检测
采用本方法对市售的36批次酱卤肉样品进行测定。结果发现,36批次样品均检出4-MEI,含量为3.1~306.6 μg/kg;2批次样品检出2-MEI,含量为21.4~23.6 μg/kg;36批次样品均未检出1-MEI。从结果分析,酱卤肉制品中可能存在超范围使用Ⅲ、Ⅳ类焦糖色的情况,也可能是由其配料带入,例如,酱油、醋等,需要结合其生产工艺进行综合分析。
3 结论
本实验建立了酱卤肉制品中3种甲基咪唑类化合物测定的超高效液相色谱串联质谱方法,样品经水提取,MCX固相萃取柱净化后上机检测。方法操作简便、灵敏度高、结果准确可靠,能一次性测定酱卤肉制品中3种甲基咪唑类化合物,可以满足监管需要,为以后酱卤肉制品中甲基咪唑类化合物分析测定以及相关国家标准的制定提供了重要参考价值。
