表现型为视神经萎缩的NDUFV1基因新移码突变
2021-06-16张智科袁慧君张水馨
张智科,袁慧君,张水馨
1中日友好医院,北京 100029;2Bascom Palmer眼科中心,美国33136;3北京市海淀区中医医院,北京100080
呼吸酶链复合物I(complex I)是线粒体内膜进行氧化磷酸化的重要结构,由37个核基因和7个线粒体基因编码。NDUFV1是编码complex I亚单元的核基因,在不同物种间高度保守,对酶的活性起关键作用。目前研究发现NDUFV1基因突变可引起complex I功能异常,是遗传性脑白质病变的重要原因[1],表现为脑白质营养不良、肌张力减低、锥体束症状、Leigh综合征等神经系统异常,并以脑部MRI中脑白质异常影像(T2高信号,T1低信号)为特征[2]。本研究报道一例新型NDUFV1基因突变,表现为儿童时期发病的双眼视神经萎缩,MRI无脑白质异常表现,并对该新型基因型、表现型进行分析。
1 资料和方法
1.1 研究对象
收集视患儿及其家人的病史及检查资料,包括父母及姐姐共4例。
1.2 方法
1.2.1 检查方法 详细询问患儿及其家人病史,进行全面眼科检查,包括视力、最佳矫正视力、裂隙灯、眼底照相、光学相干断层扫描(OCT)、视野、图形视觉诱发电位(VEP)。完善肌力、肌张力、浅感觉、深感觉等神经系统检查,及头颅核磁(MRI)检查。
1.2.2 基因检测 抽取患儿及父母外周血2 mL,提取DNA,使用二代测序进行基因检测。方法如下,采用外显子捕获试剂盒(Agilent)定位外显子区域,对这些区域使用新一代测序仪(Illumina NextSeq 500,Illumina)进行100 bp双端测序的二代测序。所得DNA序列参考人类基因组(GRCh37/UCSC)进行组装和对齐,并使用分析软件(Xome Analyzer)进行分析。对所发现的患儿及亲属的病理性变异进行Sanger测序进行验证。
1.2.3 基因致病性分析 比较基因突变位点所编码氨基酸的保守型(CLC Main Workbench软件v6.7),使用软件分析潜在致病性(PolyPhen-2软件和Mutation Taster软件)。
1.2.4 蛋白结构分析 依据所得突变构建氨基酸序列,并与野生型氨基酸序列比较。使用SWISS-MODEL在线分析工具(http://swissmodel.expasy.org/)构建野生型和突变型蛋白三维结构。
2 结果
2.1 临床表现
13岁女性患儿,近3年双眼视力下降。既往有注意力缺陷和学习障碍。父母和1个姐姐身体健康。出现症状后曾服用辅酶Q、左旋肉碱、α硫辛酸和复合维生素B。患儿神经可查体未见明显异常,双眼视力均为0.05,眼压右眼12 mmHg,左眼15 mmHg。右眼上方、鼻侧和中央视野缺损,左眼上方和颞上方视野缺损(配合欠佳,右眼光敏度-22.6 db,左眼-8.2 db)。眼底检查显示双眼视盘颞侧苍白。OCT显示双眼视神经纤维层(RNFL)缺损及神经节细胞层(RGC)萎缩(右眼114 μm,左眼52 μm)。图形视觉诱发电位(Pattern VEP)振幅降低(图1)。头颅MRI显示双侧视神经变细,未发现脑白质异常的信号(图2)。

图1 患儿眼部检查结果Fig.1 Eye examination of the child.A,B:Temporal pallor to both optic nerves;C,D:OCT showed decreased RNFL thickness;E,F:VEP showed low amplitude.RNFL:Retina nerve fiber layer.
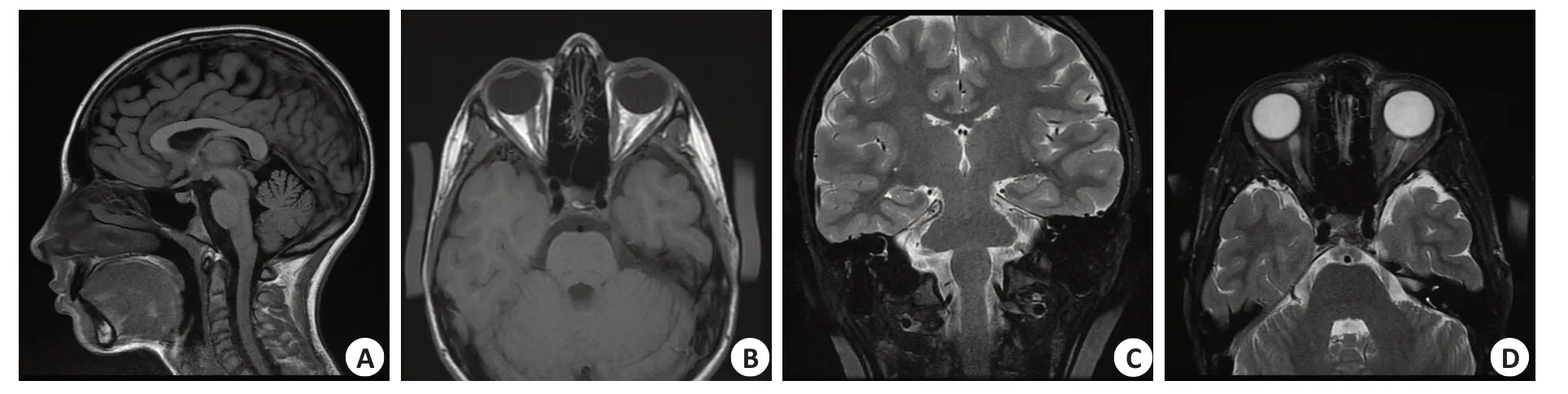
图2 患儿头颅MRI图像Fig.2 Brain MRI of the child.T1(A,B)and T2(C,D)of MRI showed no evidence of brain lesion.
治疗采用艾地苯醌125 mg 2次/d口服,1月后改为500 mg 3 次/d 口服2 月,患儿感视力稍有提高,右眼0.07,左眼0.06。服药半年后视力无明显变化遂停药。1年随访视力右眼0.07,左眼0.01。
2.2 测序结果
基因检测发现患儿存在NDUFV1 基因(NM_007103.3)外显子1 中出现杂合的移码突变(c.53_54delTG),T、G两个碱基删除使缬氨酸变为丙氨酸,引起新读码框在20 位置过早形成终止编码(p Val18AlafsX20)。同时在NDUFV1基因(NM_007103.3)内含子8 中存在杂合的点突变(c.1162+4A>C)。未发现线粒体基因突变。对父母基因测序显示c.53_54delTG来源于母亲,c.1162+4A>C来源于父亲,父母拒绝其姐姐基因检测。
2.3 突变和蛋白结构分析
在不同物种间c.53_54delTG突变及c.1162+4A>C所在位点的氨基酸具有高度保守性。使用PolyPhen-2和Mutation Taster分析提示上述两种为致病突变。
对NDUFV1 蛋白三维结构分析表明,p Val18AlafsX20引起NDUFV1第18个氨基酸之后的氨基酸序列缺失,失去了应有的黄素单核苷酸(FMN)和铁-硫簇结构,与野生型相比发生明显变化(图3)。
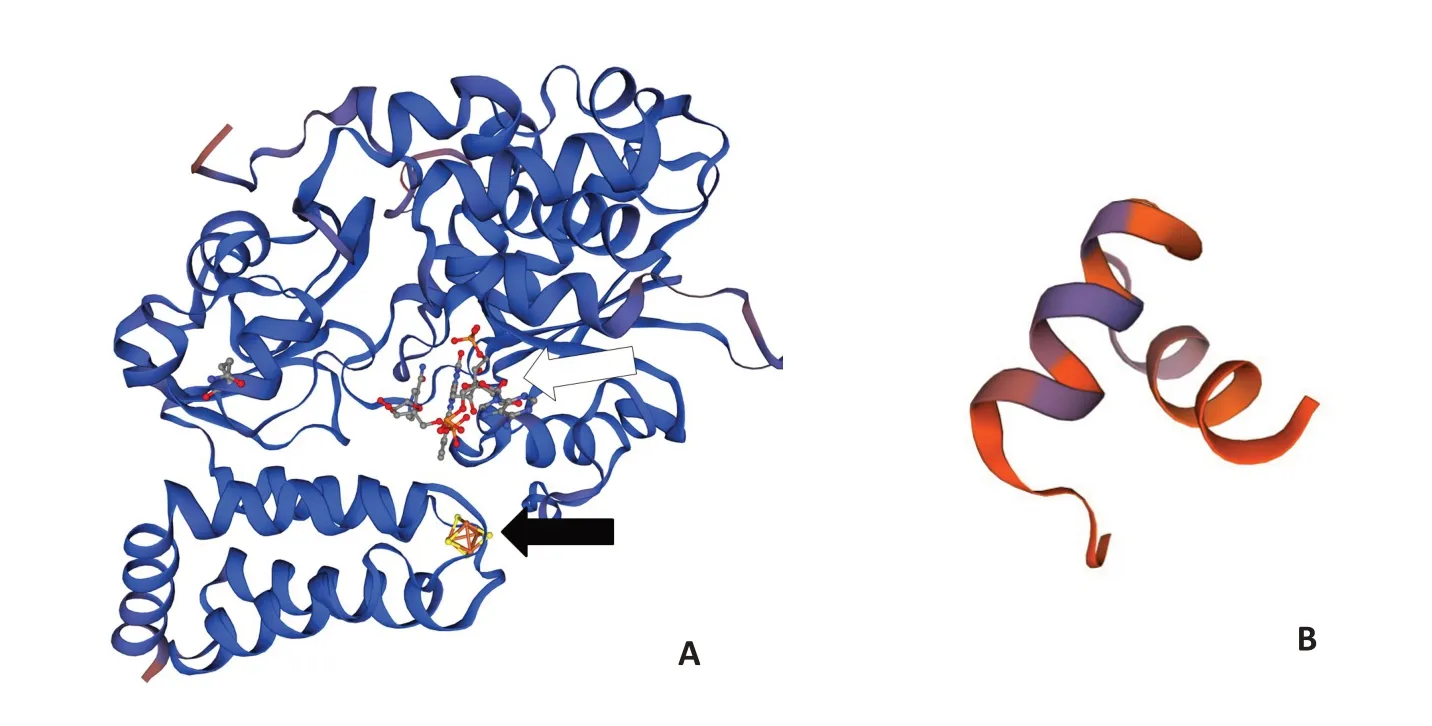
图3 野生型(A)和突变(B)NDUFV1的三维结构Fig.3 3D structure of the wild-type (A) and mutant (B) NDUFV1.Mutant NDUFV1 lost almost all main structure including flavin mononucleotide(white arrow)and Fe-S cluster(black arrow).
3 讨论
线粒体complex I是线粒体呼吸酶链的重要结构,将电子从NADH转移到辅酶Q。NDUFV1基因突变的致病性与complex I功能异常有关,主要影响下面三种功能:(1)结合FMN及NADH;(2)将电子转移到铁-硫簇;(3)维持complex I各亚单元结构的稳定性[3]。
NDUFV1基因突变多引起中枢神经系统症状,包括肌张力减低、锥体束功能异常、肌阵挛癫痫、癫痫发作、共济失调等[4],并认为是Leigh综合征的一个重要致病基因,在MRI上呈现脑白质病变表现[5-7]。Vilain等[8]发现NDUFV1基因突变个体中骨骼肌组织complex I功能明显低于对照组,肝脏组织则功能正常,表明骨骼肌症状与肌细胞中complex I功能异常有关。本研究中的NDUFV1未引起明显中枢神经系统及骨骼肌相关症状,在MRI上未出现脑白质病变征象,呈现不同的表现型。既往报道突变多为错义突变[8-9],而本研究发现突变为移码突变,表现型不同可能跟突变类型不同有关。既往研究也发现NDUFV1基因突变除表现为Leigh综合征的严重全身症状外,也可表现为学习障碍、语言发育迟缓等轻度症状,呈现多种表现型[7],表明基因型和表现型之间具有复杂的关系。
既往NDUFV1基因突变多报道为错义突变,造成FMN结合位点或铁-硫簇结构异常,影响complex I功能[10],也可引起complex I 的亚单元组装缺陷,造成complex I 数量减少[11]。而本研究所发现的新型NDUFV1基因移码突变所引起的蛋白功能异常则不同,表现为NDUFV1关键性的FMN和铁-硫簇的功能均缺失,提示新的致病机制。
本研究同时发现了NDUFV1的另一个内含子的突变。Benit等[12]曾报道一例癫痫、运动异常的婴儿具有NDUFV1 外显子5 突变(c.640G>A)、内含子8 突变(c.1192+4A>C),内含子8突变与本研究发现的c.1162+4A>C位点接近。Benit等的病例与本研究具有相似的特点,患儿分别从父母遗传两种不同NDUFV1基因突变,引起了complex I功能异常的症状,而父母未发病。内含子8点突变可引其基因剪切异常,导致外显子8表达异常及RNA的不稳定,引起complex I功能异常[12]。且该突变位点在各物种之间高度保守,因此,认为本研究发现的c.1162+4A>C是与表型相关的病理性突变。
既往没有表现为视神经萎缩的NDUFV1突变的报道。该患儿主要表现是双眼进行性的视神经萎缩,应与视神经炎鉴别。该患儿没有急性视力下降病史,进行了详细的神经查体,无肌力、肌张力、感觉异常、共济失调等其他神经系统表现,脑脊液检查无明显异常,与视神经炎表现不符。患儿MRI无脑积水表现,脑脊液压力正常,排除高颅压引起视神经萎缩。患儿无外伤、脑部肿瘤、视网膜色素变性等可引起继发性视神经萎缩的原发疾病。排除其他原因视神经萎缩后考虑为遗传性视神经疾病。最常见的遗传性视神经疾病是Leber遗传性视神经病变(LHON),是线粒体基因突变引起complex I功能异常引发的视神经表现为主的疾病。虽然该患儿表现型接近LHON,但该患儿并未发现线粒体基因的异常,反而在全外显子测序中发现了与complex I相关的核基因,即NDUFV1的异常。对NDUFV1基因的进一步研究有助于揭示complex I异常与视神经萎缩间的病生理机制。有研究发现存在线粒体基因ND1突变及NDUFV1 基因突变共同存在的家系,引起complex I功能和数量的改变,提示线粒体基因ND1与NDUFV1共同作用引起中枢神经系统症状[11]。
目前缺乏NDUFV1基因突变相关疾病的有效治疗方式。既往报道该基因突变患儿多于出生数月至3岁早夭,预后较差,目前治疗原理上是通过补充呼吸酶链必不可少的核黄素,期望改善complex I 功能[3]。Benit等[12]报道使用补充B族维生素治疗取得了一定的效果,也有报道采用生酮饮食改善了症状[13]。本研究通过补充艾地苯醌在初期一定程度改善了症状,对该类疾病的治疗有一定借鉴意义。
综上所述,本研究报道了一例新NDUFV1基因突变,表现为儿童发病的视神经萎缩,其表现型与既往NDUFV1基因突变不同。该NDUFV1基因突变引起complex I 亚单元蛋白结构异常,但该异常引起complex I功能的变化需进一步研究,并且有待进一步研究发现其引起视神经萎缩却不引起中枢神经系统异常的机制。
