vicK基因ATP结合位点对变异链球菌生物学功能的影响
2021-06-03黄珊珊宋秀宇徐巧丽饶慧华张加勤
黄珊珊,宋秀宇,徐巧丽,饶慧华,张加勤
变异链球菌(Streptococcusmutans,S.mutans)是口腔主要致龋菌之一,至今无特效药根治龋齿。变异链球菌感染口腔后粘附牙齿表面形成生物膜,并在其保护下分解糖类,产生酸代谢产物,引发龋齿[1]。而在致龋的过程中,变异链球菌需克服各种生存环境的改变生长,这主要依赖原核生物信号传导双组份系统(two-component system,TCS)的调控作用[2]。TCS可参与调节细菌的多种生物学作用,如细胞壁形成、粘附、毒力、氧化应激反应等[3]。S.mutansUA159可编码14 种TCSs,VicRK双组分系统为其中之一,与细菌毒力相关,VicK为组氨酸激酶(Histidine Kinase,HK),VicR为可与DNA结合的转录因子应答调节蛋白(Response Regulator,RR)[4]。
HATPase_c结构域(CA区)是VicK中具有ATP酶活性的特征性区域,显示激酶活性,是细菌激酶磷酸化的始动环节。以往人们对VicK的研究大多是敲除整个vicK基因,没有单独对HATPase_c结构域的ATP激酶活性进行研究。为研究vicK基因HATPase_c结构域中ATP结合位点对变异链球菌的影响,本研究采用同源重组的方法敲除vicK基因ATP结合位点核心区域(命名为ΔvicKATP基因),验证其蛋白ATP激酶活性,构建补偿菌株,观察vicK基因ATP结合位点对变异链球菌生长、产酸耐酸、胞外多糖(exopolysaccharides,EPS)和生物膜形成能力等生物学功能的影响。
1 材料和方法
1.1菌株和质粒 变异链球菌UA159由第四军医大馈赠;质粒pCrePA和pIB107由堪萨斯大学医学中心 Biswas Indranil 教授惠赠;pET-His、E.coliDH5、E.coliBL21(DE3)、pET-21(b)、pDLP为前期实验构建。

1.3实验步骤
1.3.1vicK基因HATPase_c结构域预测 利用expasy、phyre2和软件TargetATPsite[5]预测HATPase_c结构域和其中的ATP结合位点,设计引物敲除vicK基因HATPase_c结构域ATP结合位点的核心区域。
1.3.2vicKATP突变同源重组载体构建 以变异链球菌UA159基因组DNA为模板,引物见表1,PCR扩增vicK-X1-267片段,将vicK-X1-267片段和pET-His载体分别用BamHI/NheI酶切,T4 DNA连接酶建立连接体系,转化E.coliDH5感受态细胞,氨苄西林(Ampicillin,Amp)(100 g/mL)筛选阳性菌落,37 ℃过夜培养并抽提质粒获得重组载体pET-His-vicK-X1-267(pK1)。利用quickchange primer design设计引物,用quick change法构建vicKATP基因突变质粒,同时引入SmalI平末端酶切位点。quick change法建立反应体系:templet(pK1)1 μL(50 ng/μL),5×FastPfu reaction buffer 10 μL,2.5 mmol/L dNTPs 1 μL,FastPfu 0.5 μL,qF1、qR1各2 μL(10 μmol/L),加灭菌水至50 μL。反应条件:95 ℃预变性3 min;95 ℃变性45 s,55~60 ℃退火45 s,68 ℃延伸15 min,95 ℃变性,17 个循环;72 ℃延伸10 min,12 ℃永久。DpnI处理PCR产物,50 μL PCR产物中加入0.5 μLDpnI,37 ℃水浴5 h。由于模板DNA上有甲基化位点,而用PCR新扩增出来的DNA无甲基化修饰,DpnI可识别DNA序列上的甲基化位点,切断野生型模板DNA,留下突变DNA。测序验证成功的载体pET-His-vicKATP命名为pK2。SmalI酶切pK2,以质粒pIB107为模板,PCR扩增kan基因盒(lox71-kan-lox66),用PfIu DNA polymeras将lox71-kan-lox66片段补平,连接,转化。Amp(100 g/mL)和卡那霉素(Kanamycin,Kan)(50 g/mL)筛选阳性菌落,获得同源重组载体pK2:loxPkan(pK3),经PCR、酶切及测序鉴定成功。

表1 PCR扩增引物
1.3.3UA159 SΔvicKATP突变株构建 参照文献[6],将pK3经BamHI线性化后,CSP催化转化UA159,并接种于含Kan(300 g/mL)的Todd-Hewitt Broth plus 0.2%Yeast Extract(THY)固体培养基37 ℃培养24 h,筛选阳性克隆菌落(SΔvicKATP::kan)。同法将热敏质粒pCrePA转化SΔvicKATP::kan突变株,pCrePA含emr基因,接种含红霉素(Erythromycin,Em)(10 g/mL)的THY固体培养基,30 ℃ 48 h筛选阳性菌落,利用Cre酶识别loxP位点剔除kan基因。转种37 ℃培养基过夜使pCrePA质粒丢失。挑取单个菌落平行接种300 g/mL Kan、10 g/mL Em和无抗性THY固体培养基,37 ℃培养24 h。筛选Kanr和Emr平板不生长,无抗性平板上生长的菌落,即无标记SΔvicKATP突变株。测序鉴定。
1.3.4UA159vicKATP补偿株构建 以UA159基因组DNA为模版,PCR扩增vicK基因开放阅读框。vicK片段和穿梭表达质粒pDLP分别用NdeI/KpnI酶切,连接,转化,Kan(50 g/mL)LB平板筛选阳性菌落。所获质粒经PCR和酶切鉴定成功即为补偿质粒pDLP-vicK(pK4)。补偿质粒转化SΔvicKATP突变株,方法和步骤同上所述,含300 μg/mL KanrTHY平板筛选并获得vicKATP补偿株为SvicK。
1.3.5RT-PCR验证vicK基因转录 提取UA159、SΔvicKATP突变株和SvicK补偿株RNA,合成第一链cDNA,PCR扩增cDNA的vicK产物。验证SΔvicKATP突变株和SvicK补偿株的vicK基因转录活性。
1.3.6vicK基因HATPase_c结构域缺失蛋白表达纯化和ATP酶活性检测 分别以UA159和SΔvicKATP突变株基因组DNA为模版,PCR扩增vicK基因开放阅读框。BamHI和HindIII双酶切表达载体pET-21(b)、vicK基因和vicKATP基因片段,连接,转化,Ampr LB平板筛选阳性菌落。所获质粒经PCR和酶切鉴定成功即为表达载体pET-21(b)-vicK和pET-21(b)-ΔvicKATP,转化感受态细胞E.coliBL21(DE3)获得pET-21(b)-vicK-BL21和pET-21(b)-ΔvicKATP-BL21,IPTG诱导蛋白表达,Ni-NTA SefinoseTM纯化蛋白。目的蛋白经SDS-PAGE电泳分析初步鉴定,-80 ℃保存备用。
1.3.7细菌培养 生物学功能实验均将UA159、SΔvicKATP突变株和SvicK补偿株在37 ℃、5% CO2条件下静置过夜培养。
1.3.8生长速率 将菌株分别以1∶100比例接种50 mL THY液体培养基,37 ℃静置培养。每隔1 h读取OD600值,对数生长期(OD600≈0.5)每隔30 min读取1次数值。重复3次实验取平均值,记录数值并作曲线图。
1.3.9产酸实验 将菌株分别以1∶20比例接种THY培养基,37 ℃静置培养。每隔1 h测量pH值,分别为0、1、2、3、4、5、6 h。重复3次实验取平均值,记录数值并绘制曲线图。
1.3.10酸耐受实验 分别取菌株1 mL,生理盐水洗涤。加200 μL 0.05 mol/L甘氨酸-盐酸缓冲液(pH2.8),分别反应60 min、90 min,各取100 μL 菌液梯度稀释铺板。置37 ℃培养48 h。菌落计数,重复3 次实验取平均值,并绘图。
1.3.11生物膜培养 将菌株转种到THY培养基中培养至对数生长期(OD600≈0.5),以1∶100比例接种于50 mL含0.5%蔗糖的THY培养基,取1 mL 稀释物注入无菌的十二孔平底培养板中,并分别放入一张无菌盖玻片,37 ℃ CO2培养箱静置培养48 h,洗涤培养板去除未粘附细菌,风干,每孔加入250 μL 0.1%结晶紫溶液覆盖孔底,室温染色5 min,水洗多余染液并风干,镜下观察生物膜。1 mL(乙醇∶丙酮=8∶2)的萃取液萃取生物膜,酶标仪检测OD570吸光度值。
1.3.12扫描电镜(SEM)观察生物膜及EPS形成情况 生物膜培养后,取出硅片,PBS吹打洗涤细菌,除去表面生物膜的残留杂质,2.5%戊二醛固定10~20 min,30%、50%、70%、90%、100%乙醇各10~20 min干燥杯中脱水,再从乙醇逐步过渡到叔丁醇,干燥杯4 ℃贮存,电镜室冷冻干燥,喷金后电镜观察。
1.4统计学分析 数据采用Graphpad Prism 8.0软件绘图分析,应用双因素方差分析(two-way ANOVA)比较实验组和对照组ATP激酶活性、产酸、酸耐受差异,应用单因素方差分析非参数检验(Kruskal-Wallis test)比较实验组和对照组生物膜形成差异。检验水准α=0.05。
2 结 果
2.1vicK基因HATPase_c结构域预测vicK基因HATPase_c结构域和其ATP结合位点如图1所示。设计引物对ATP结合位点核心区域396-402 aa进行缺失突变,vicK基因HATPase_c结构域和其ATP结合位点位置推断突变位置不影响vicK基因其他功能结构域的表达。
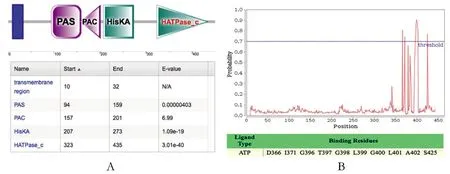
A:HATPase_c结构域预测;B:ATP结合位点预测
2.2UA159 SΔvicKATP突变株构建与鉴定 构建成功的同源重组载体pK3经BamHI线性化后转化UA159,在Kanr平板上生长说明lox71-kan-lox66已经成功整合到UA159基因组。热敏质粒pCrePA转化ΔvicKATP::kan突变株,利用Cre酶识别loxP位点可剔除kan基因。37 ℃使pCrePA质粒丢失。突变株在Kanr和EmrTHY平板上不生长,只在无抗性THY平板上生长,验证kan基因和pCrePA质粒均成功去除,成功构建无标记SΔvicKATP突变株。SΔvicKATP突变株测序结果显示vicK基因的ATP结合位点序列已缺失,只存留38 bp的loxP位点(图2)。
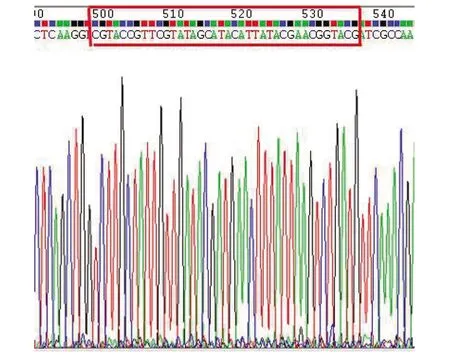
红色方框内为删除kan基因后残留38 bp的loxP位点
2.3UA159vicK补偿株构建与鉴定 将构建成功的补偿质粒pDLP-vicK(pK4)转化vicKATP突变株,Kanr的THY平板筛选获得vicK补偿株。由于SΔvicKATP突变株删除了ATP结合位点核心区域21 bp,引入lox71-kan-lox66,而Cre重组酶识别loxP位点删除kan基因后残留两端38 bp的loxP位点,用PCR扩增vicK基因和突变基因产物大小区别不大,无法区分验证突变株和补偿株,且无法确定补偿vicK基因是否有转录活性。因此,对阳性转化SvicK补偿株和SΔvicKATP突变株vicK基因转录情况用RT-PCR验证。结果如下图所示,SΔvicKATP突变株RT-PCR产物电泳结果无相关条带出现,进一步说明SΔvicKATP突变株构建成功,SvicK补偿株vicK基因有转录活性(图3)。
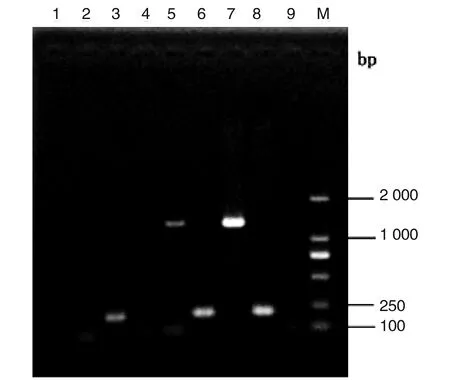
1:突变株SΔvicKATP RNA的RT-PCR扩增vicK产物;2:突变株SΔvicKATP cDNA 的RT-PCR扩增vicK产物;3:突变株SΔvicKATP cDNA 的RT-PCR扩增gyrA内参产物;4:补偿株SvicK RNA的RT-PCR扩增vicK产物;5:补偿株SvicK cDNA的RT-PCR扩增vicK产物;6:补偿株SvicK cDNA的RT-PCR扩增gyrA内参产物;7:UA159野生株基因组的PCR扩增vicK产物;8:UA159野生株基因组的PCR扩增gyrA内参产物;9:阴性对照;M:DNA Marker DL2000
2.4vicK基因HATPase_c结构域缺失蛋白表达纯化和ATP酶活性检测 经IPTG诱导表达目的蛋白,ΔVicKATP蛋白为45.7 kDa,VicK蛋白为50 kDa。Ni-NTA SefinoseTM纯化蛋白,imidazole浓度400 mmol/L洗脱目的蛋白(图4)。检测ΔVicKATP蛋白和VicK蛋白的ATP激酶活性,绘制标准曲线(图5A)。结果提示随ATP浓度增加,发光强度增加。蛋白ATP酶活性越高,结合ATP能力越强,导致游离ATP减少,发光强度降低,即蛋白ATP酶活性与发光强度成反比。随着两种蛋白摩尔浓度增加,VicK蛋白发光强度呈明显降低趋势,而ΔVicKATP蛋白发光强度无明显变化,差异有统计学意义(FVicK-ΔVicKATP=22.87,P<0.05)(图5B)。结果显示,VicK蛋白的ATP激酶活性明显高于ΔVicKATP蛋白,ΔVicKATP蛋白几乎失去ATP激酶活性。验证了SΔvicKATP突变株构建成功,缺失了ATP的结合能力。
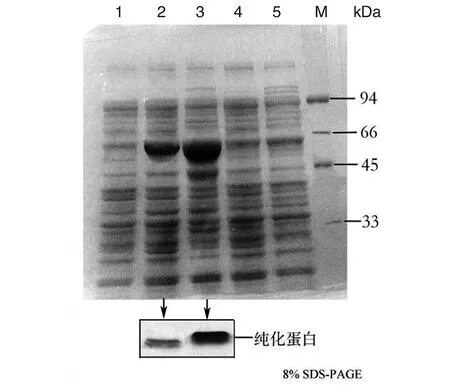
1:对照株E.coli BL21(DE3)裂解上清液;2: pET-21(b)-ΔvicKATP-BL21裂解上清液及纯化蛋白;3: pET-21(b)-vicK-BL21裂解上清液及纯化蛋白;4:pET-21(b)-ΔvicKATP-BL21裂解沉淀物;5:pET-21(b)-vicK-BL21裂解沉淀物;M:蛋白质marker
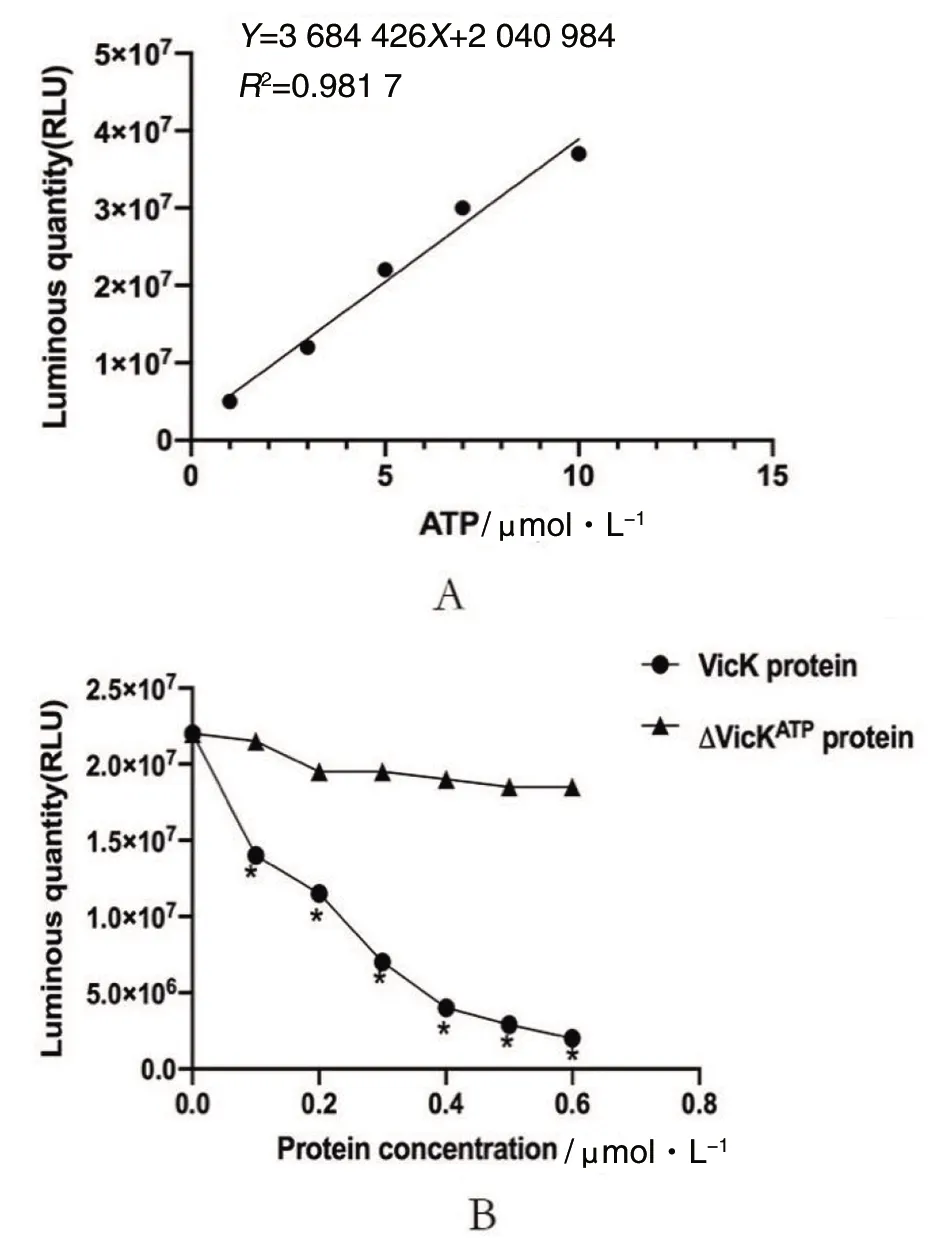
A:标准曲线;B:不同摩尔浓度ΔVicKATP蛋白和VicK蛋白ATP激酶活性;*P<0.05,compared with ΔVicKATP protein
2.5生长曲线 每隔60 min测量OD600。与野生株UA159比较,SΔvicKATP突变株生长较为缓慢,但稳定期基本可达到同一水平,而SvicK补偿株基本恢复至野生株的生长水平,见图6。
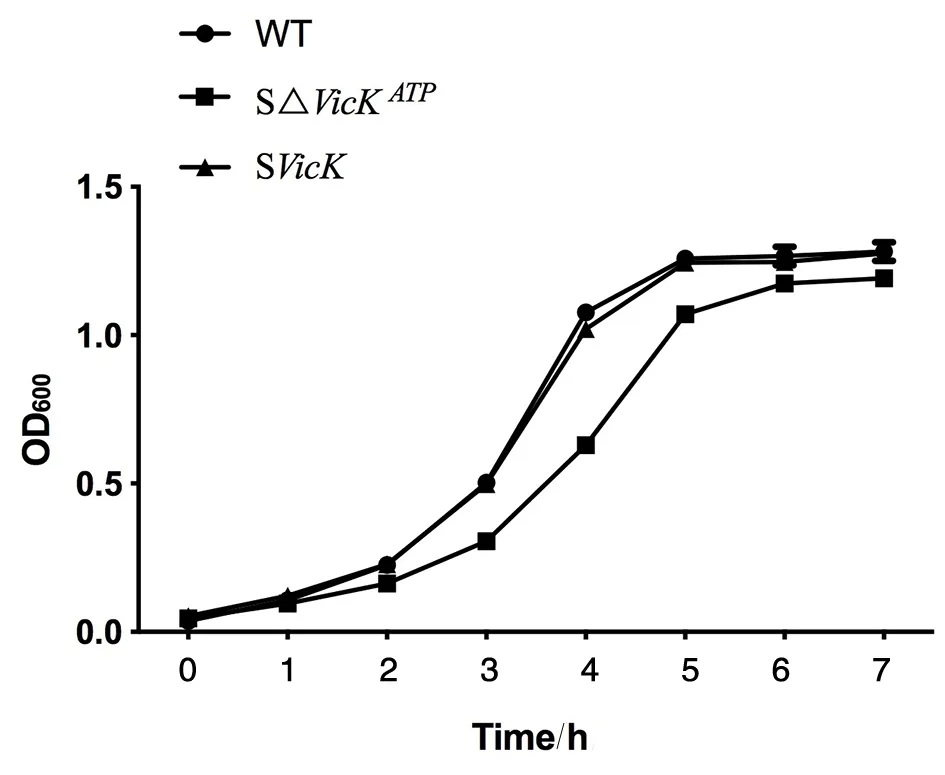
图6 UA159、SΔvicKATP突变株和SvicK补偿株的生长曲线
2.6产酸实验 相比野生株,随时间增加SΔvicKATP突变株PH值下降更缓慢,即突变株产酸量减少,差异有统计学意义(q1、2、3、4、5、6:WT-SΔvicKATP=7、8.875、18.35、40.83、99.03、10,P<0.05)。SvicK补偿株pH值下降也较野生株缓慢,产酸量稍减少,部分差异有统计学意义(q2、3、4、5:WT-SvicK=13、61、116、164,P<0.05),但没有突变株明显,说明ATP结合位点突变使菌株产酸量减少,补偿株不能完全恢复到野生株水平(图7)。
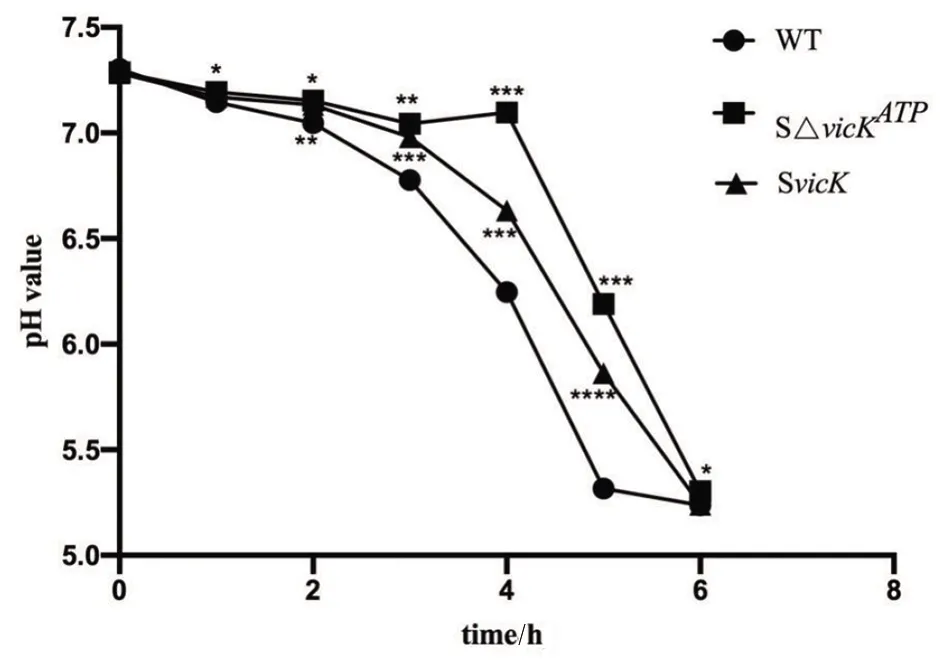
*P<0.05,**P<0.01,***P<0.005,****P<0.001,compared with WT
2.7酸耐受实验 在致死性酸性环境中(pH 2.8),随着酸处理时间延长,UA159、SΔvicKATP突变株、SvicK补偿株生存率呈下降趋势,但与野生株相比,SΔvicKATP突变株反而下降的更少,差异有统计学意义(q60:WT-90:WT=45.20,q60:WT-90:SΔvicKATP=14.14,q60:WT-90: SvicK=34.96,P<0.05),提示突变株耐酸性增高。90 min时,补偿株和野生株差异有统计学意义(q90:WT-SvicK=23.03,P<0.05),提示随酸处理时间增加,ATP结合位点突变对酸耐受性增加(图8)。
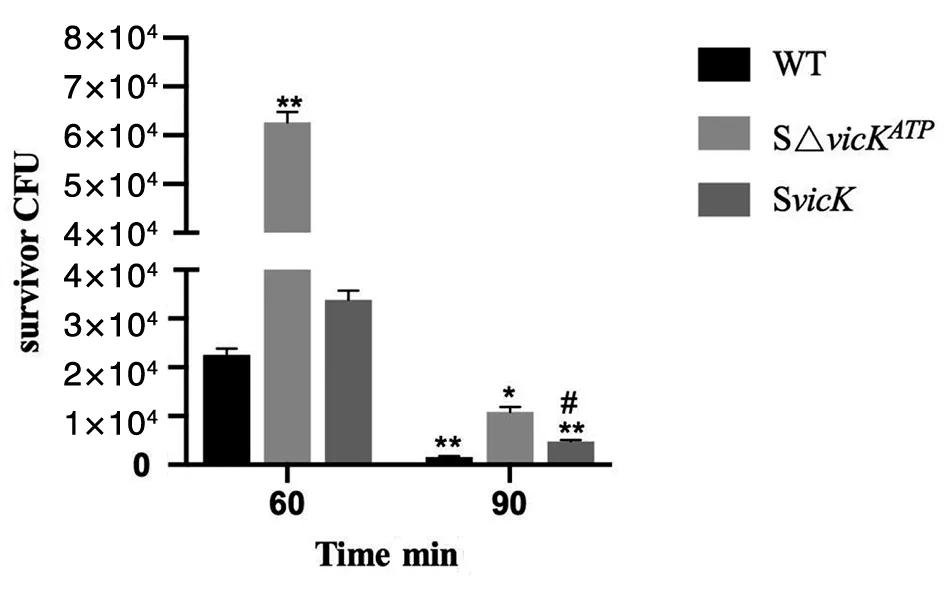
*P<0.05,**P<0.01,compared with WT60;#P<0.05, compared with WT90
2.8vicK基因ATP结合位点突变对变异链球菌生物膜及EPS形成的影响 与野生株相比,SΔvicKATP突变株生物膜形成明显减少,粘附力降低,且致密性降低,补偿株SvicK生物膜形成能力基本与野生株相当(图9)。用配制的溶液萃取生物膜,将萃取液于OD570检测,结果与结晶紫染色一致:突变株与野生株比较,差异有统计学意义(Z=2.695,P<0.05),说明SΔvicKATP突变株使变异链球菌生物膜形成能力降低。补偿株与野生株比较差异无统计学意义,从图9中看出补偿株起到了部分补偿作用,但还是有一定差异。电镜SEM结果观察到:SΔvicKATP突变株玻片上的生物膜比野生株薄,与生物膜培养结晶紫染色结果一致,细菌量比野生株少,EPS生成量相比野生株明显减少,补偿株几乎恢复到野生株水平(图10)。
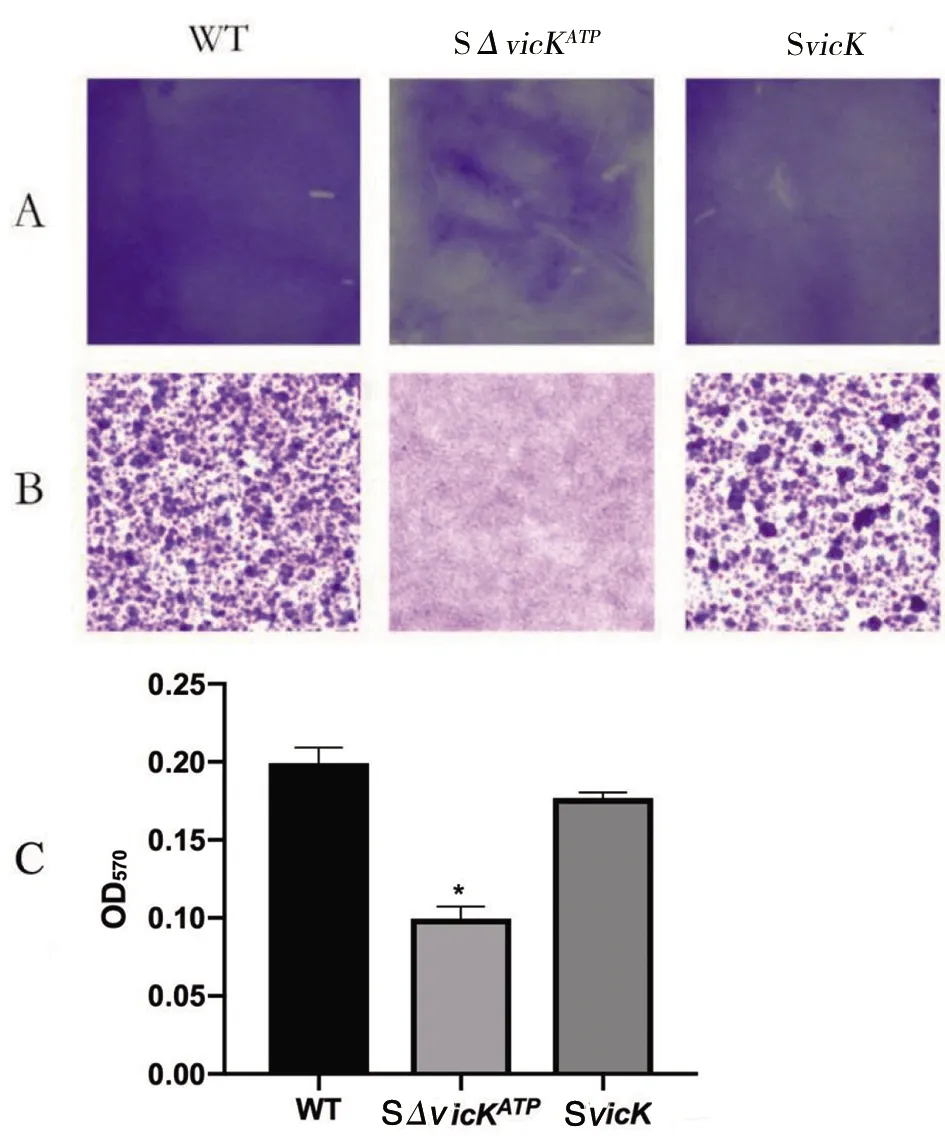
A:生物膜形成;B:光学显微镜下生物膜形态(100倍);C:生物膜萃取液OD570值;*P<0.05,compared with WT

A:50 000倍;B:5 000倍
3 讨 论
TCS由位于膜上的组氨酸激酶受体HK和位于胞内的调节子RR组成[7]。HK感受外界信号并发生自磷酸化,将磷酸基团转移到RR上将其磷酸化,调控下游目的基因表达[3,8]。HK由N端传感器复合体和C端催化中心组成。N端包括胞外感受区、跨膜区(TM)和胞内感受区(HAMP、PAS)[9]。C端包括DHp和HATPase_c结构域(CA区)。HATPase_c结构域具有催化活性并含有 ATP 结合位点,识别结合ATP,并呈递磷酸基团给DHp区的His217,使其发生自身磷酸化,是细菌激酶磷酸化的始动环节[10]。细菌HATPase_c结构域在人体中没有发现,近年来已成为抗菌药物研究的重要靶点[11]。研究表明双组分系统主要存在于细菌、一些低等真核生物和一些原生动物中,在人体真核细胞中却不存在[12],因此可通过研究双组分系统蛋白毒力调节机制,寻找变异链球菌双组分系统中的药物新靶点,特异高效地抑制病原菌而对宿主不造成影响。
变异链球菌中的双组分VicRK系统的vicK基因,其编码的组氨酸激酶VicK感受外界刺激信号并传递给应答调节蛋白VicR,开始信号通路转导,从而调节相关蛋白的转录翻译[13]。因此,vicK基因在变异链球菌调节毒力机制作用的研究至关重要。研究表明,vicK基因对变异链球菌的生长、产酸、耐酸、粘附力、氧化应激、生物膜和糖酵解均有调控作用,对产生EPS研究较少,EPS对细菌生物膜形成和毒力均有重要作用[2,4,14-16]。这些大多是敲除整个vicK基因的研究结果。但VicRK是复杂的双组分调控系统,VicK不仅有组氨酸激酶活性,还有磷酸酶和磷酸转移酶活性。为研究vicK基因HATPase_c结构域中ATP结合位点激酶磷酸化功能对变异链球菌的影响,本研究通过敲除vicK基因ATP结合位点,观察vicK基因ATP结合位点对变异链球菌生物学功能的影响。研究表明,VicK蛋白的保守氨基酸残基P222、T221和A439有磷酸酶活性,其中P222、T221点突变,VicK蛋白失去磷酸酶活性,A439位点缺失突变,VicK蛋白磷酸酶活性显著降低;A439和W443有磷酸转移酶活性,突变后VicK蛋白磷酸转移酶活性显著降低10。本研究中对HATPase_c结构域中ATP结合位点核心区域396-402 aa进行缺失突变,突变位点不涉及已知磷酸酶和磷酸转移酶活性位点,保留了磷酸酶和磷酸转移酶活性。蛋白结构预测分析发现,VicK蛋白氨基酸全长450 aa,HATPase_c结构域位于C端323-435 aa,突变位置不影响vicK基因其他功能结构域的表达。
为研究vicK基因ATP结合位点对变异链球菌生物学功能的影响,本研究用基因克隆和quick change法成功构建了vicKATP基因突变同源重组表达载体。利用Cre-loxP系统和同源重组法将ATP结合位点突变的目的基因整合到变异链球菌的基因组中,稳定表达突变目的基因,成功构建SΔvicKATP突变株。利用Cre酶识别并删除引入的lox71-kan-lox66抗性基因盒,构建无标记的SΔvicKATP突变株。同时,克隆vicK片段,采用穿梭表达质粒pDLP转化SΔvicKATP突变株,成功构建具有vicK基因转录活性的补偿株SvicK。成功构建VicKATP蛋白表达株,表达纯化蛋白,并检测其几乎失去ATP酶活性,验证了SΔvicKATP突变株构建成功,缺失了ATP的结合能力。通过描绘生长曲线、产酸、酸耐受、EPS和生物膜等生物学功能实验,研究vicK基因ATP结合位点对变异链球菌的影响。研究结果显示:与野生株UA159比较,SΔvicKATP突变株生长和产酸都受到抑制,酸耐受性增强,SvicK补偿株不能完全恢复到野生株水平;生物膜通过结晶紫染色、电镜SEM观察,SΔvicKATP突变株生物膜形成明显减少,粘附力降低,且致密性降低,生物膜形成能力降低,补偿株SvicK生物膜形成能力基本与野生株相当,其中SEM还观察到SΔvicKATP突变株EPS生成量相比野生株明显减少。实验结果与S.mutansUA159的vicK基因全敲除结果大致相同,揭示vicK基因HATPase_c结构域中ATP结合位点对变异链球菌生物学功能和毒力机制有重要影响,具体机制待进一步研究。
变异链球菌vicKATP基因的成功敲除及生物学功能研究,为进一步研究变异链球菌vicK基因ATP结合位点表达调控毒力机制提供了无标记基因缺陷株模型和实验依据,为研发抗菌药物奠定基础。
利益冲突:无
