基于有效基准特征图谱质量表征的卷丹饮片质量评价研究
2021-05-17范圆圆李林福李亚琪黄玉娟高园园舒一崧孔静潘锋孙仁弟黄广伟姜艳艳石任兵
范圆圆 李林福 李亚琪 黄玉娟 高园园 舒一崧 孔静 潘锋 孙仁弟 黄广伟 姜艳艳 石任兵
卷丹为百合科植物卷丹LilumlancifoliumThunb.)的干燥肉质鳞叶,为2020版《中国药典》规定的三种药用百合来源之一。其药食两用,性寒味甘微苦,入心、肺经。有养阴润肺,清心安神的功效。临床上常用于阴虚燥咳,劳嗽咳血,虚烦惊悸等症[1]。现代药效作用研究表明,卷丹具有明显的降血糖作用[2-3]。
卷丹中主要含有酚类、甾体皂苷类、多糖类等主要化学类型,亦含有生物碱、磷脂、氨基酸以及微量元素等成分[4]。课题组前期对卷丹中皂苷类及酚类有效部位化学成分进行了系统研究,并分别建立了皂苷类及酚类指标性成分含量测定方法及HPLC特征图谱[5-7]。近年来,对百合降糖作用的研究主要集中在百合多糖,同时也有文献根据体外实验推测百合甾体皂苷具有潜在的降糖作用[8-12]。课题组前期在基于降糖作用对百合进行药物体系研究时,发现百合中的酚类成分和甾体皂苷类成分为其药物体系架构的主要有效物质基础,并建立了同时测定百合酚类及甾体皂苷类成分含量测定方法及特征图谱[13]。笔者基于本课题组前期研究积淀[14-15],建立卷丹有效基准特征图谱质量表征方法,其中涵盖了卷丹两类主要化学类型酚类及甾体皂苷类,并对模拟基准的准确性进行佐证及质量评价应用与验证。以精准、有效、便捷地评价卷丹饮片的质量,为卷丹的应用提供科学依据,也为有关药物的质量有效评价与控制提供借鉴。
1 材料
1.1 仪器
Waters e2695自动进样高效液相色谱仪,2998型光电二极管矩阵检测器(美国沃特世公司),Empower工作站;Sartorius BT 125D型1/100 000电子分析天平;KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司);数显恒温水浴锅SYG-4(常州朗越仪器有限公司)。
1.2 试剂与试药
王百合苷C对照品(批号:DST201027-076),王百合苷A对照品(批号:DST201029-107),王百合苷E对照品(批号:DST201027-075),王百合苷B对照品(批号:DST200602-071),均购自成都德思特生物技术有限公司,对照品纯度均≥98%;卷丹皂苷[(25R)-3β,17α-二羟基-5α-螺甾烷-6-酮-3-O-α-L-鼠李糖基-(1→2)-β-D-葡萄糖苷,批号:18080801],购自成都普菲德生物技术有限公司,对照品纯度≥98%;色谱级乙腈购自Fisher Scientific公司;娃哈哈纯净水,购自杭州娃哈哈集团有限公司;甲醇,购自天津市大茂化学试剂厂;磷酸,购自北京化工厂。16批卷丹饮片购自全国各地,检验报告均符合2015版《中华人民共和国药典》相关规定,经北京中医药大学刘春生和王学勇教授鉴定为百合科植物卷丹的干燥肉质鳞叶。卷丹饮片的产地、批号、生产商及采购地见表1,饮片样品现存于北京中医药大学中药学院中药化学系。
2 方法与结果
2.1 有效基准特征图谱质量表征方法的建立
2.1.1 混合对照品溶液的制备 精密称量9.50 mg王百合苷C对照品,用80%甲醇溶解并定容至5 mL棕色容量瓶中,即得王百合苷C对照品母液。精密称量9.35 mg王百合苷E对照品,用80%甲醇溶解并定容至5 mL棕色容量瓶中,即得王百合苷E对照品母液。精密称量32.25 mg王百合苷A对照品,用80%甲醇溶解并定容至5 mL棕色容量瓶中,即得王百合苷A对照品母液。精密称量46.50 mg王百合苷B对照品,用80%甲醇溶解并定容至5 mL棕色容量瓶中,即得王百合苷B对照品母液。精密称量14.75 mg卷丹皂苷对照品,用80%甲醇溶解并定容至5 mL棕色容量瓶中,即得卷丹皂苷对照品母液。分别吸取王百合苷C及王百合苷E对照品母液0.1、0.2、0.3、0.4及0.5 mL,王百合苷A对照品母液0.4、0.5、0.6、0.7及0.8 mL,王百合苷B对照品母液0.3、0.35、0.4、0.45及0.5 mL,卷丹皂苷0.4、0.6、0.8、1.0及1.2 mL至5 mL棕色容量瓶中,用80%甲醇定容,即得5个浓度梯度的混合对照品溶液。
2.1.2 供试品溶液的制备 取卷丹饮片基准粉末(S16)及待评价饮片粉末(过4号筛)2 g,精密称定,置具塞锥形瓶中,精密加入100倍量80%甲醇,加热回流60 分钟,滤液蒸干,残渣加80%甲醇溶解并完全转移至5 mL棕色容量瓶中,再用80%甲醇定容至刻度线,摇匀,0.45 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
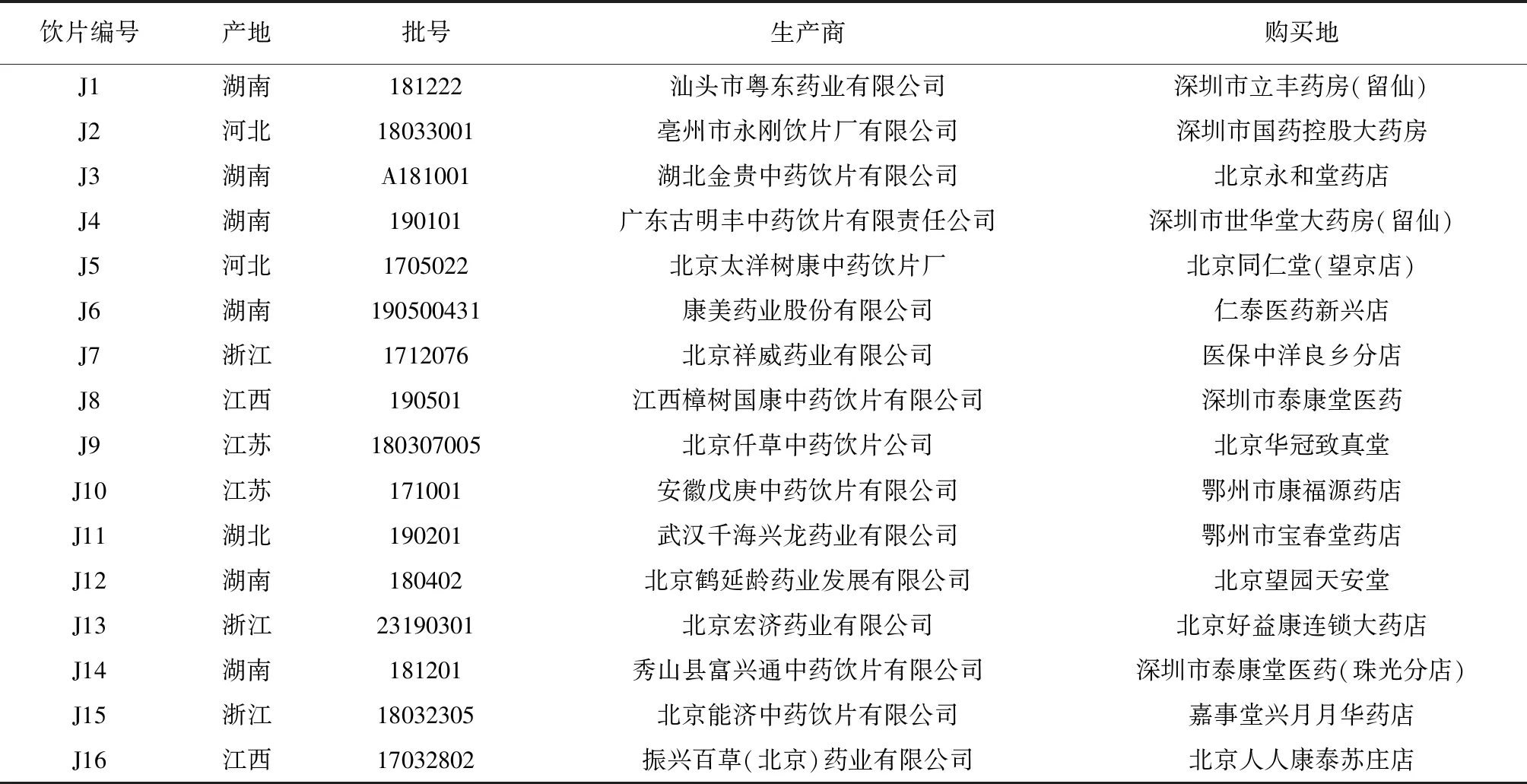
表1 16批卷丹饮片信息
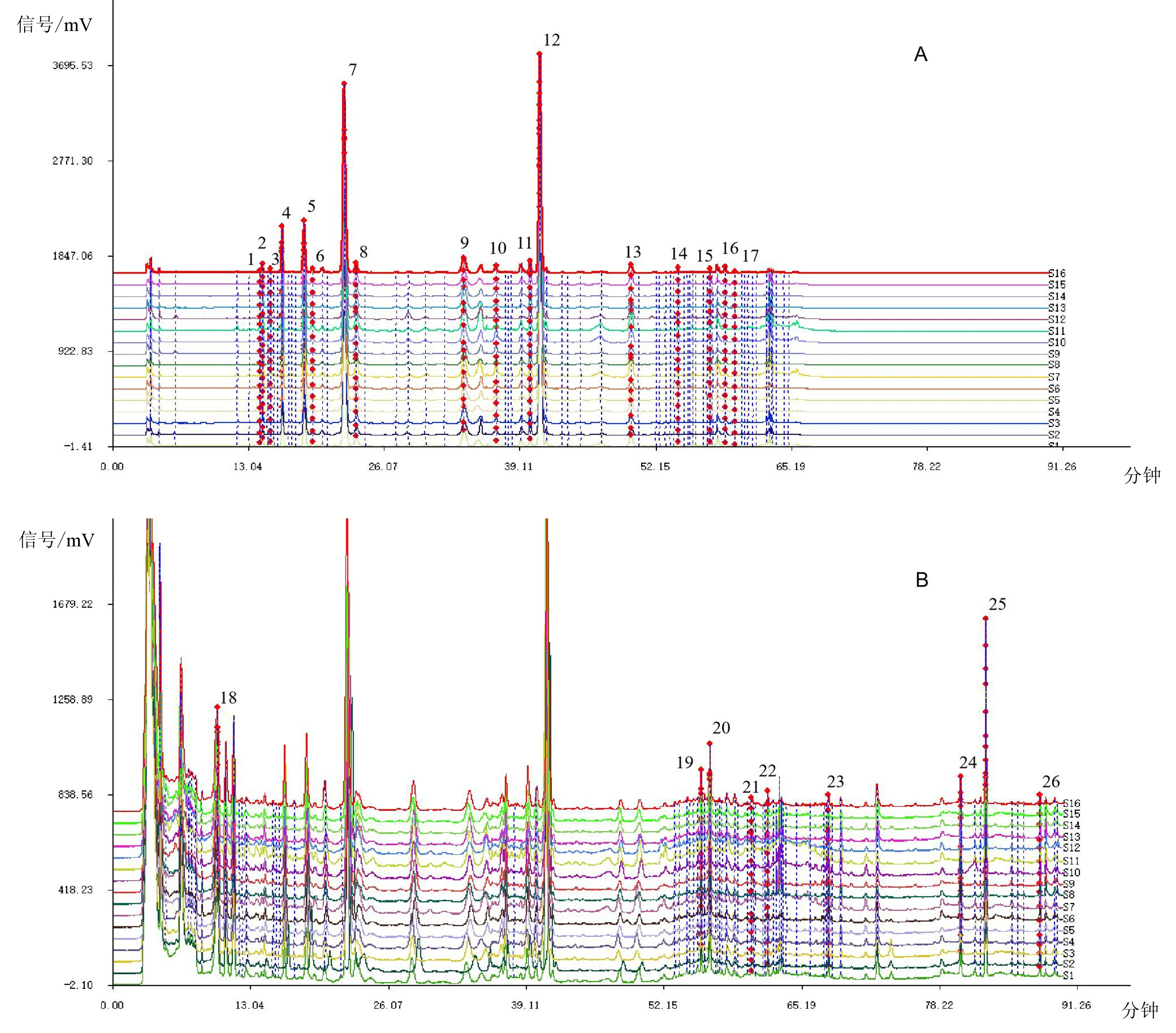
2.1.3 色谱检测条件 Waters Xbridge Shield RP C18(4.6×250 mm,5 μm)色谱柱;流动相乙腈(A)-0.05%磷酸水(B),洗脱梯度(0~10 分钟,5%~14%A;10~30 分钟,14%A;30~31 分钟,14%~21%A;31~45 分钟,21%A;45~55 分钟,21%~36%A;55~63 分钟,36%~66.3%A;63~70 分钟,66.3%A;70~83 分钟,66.3%~100%A;83~90 分钟,100%A),流速0.8 mL/min,柱温30℃,酚类成分检测波长为330 nm,甾体皂苷类成分检测波长为205 nm,进样20 μL。卷丹饮片(S16)样品、混合对照品在以上色谱检测条件下的HPLC色谱图,见图1。
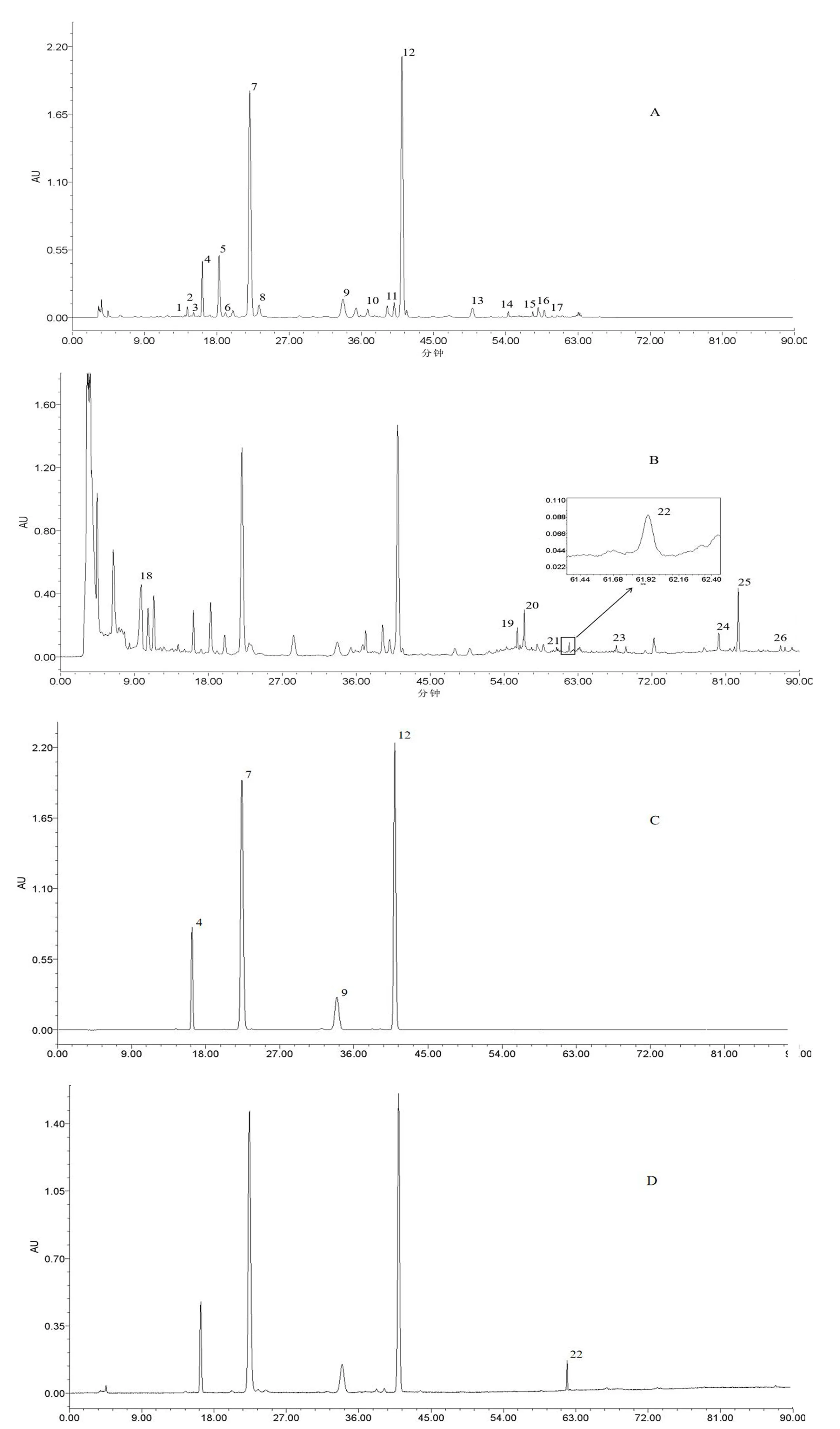
注:A供试品(330 nm);B供试品(205 nm);C混合对照品(330 nm);D混合对照品(205 nm);4王百合苷C;7王百合苷A;9王百合苷E;12王百合苷B;22卷丹皂苷;1-17酚类成分;18-26甾体皂苷类成分。
2.1.4 有效基准特征图谱方法学考察 (1)精密度考察:按“2.1.2”制备S16供试品溶液,按“2.1.3”设定检测条件,连续进样6次,以王百合苷C为参照峰,计算25个色谱峰与参照峰的相对保留时间及相对峰面积。计算结果显示,25个色谱峰与参照峰的相对保留时间和相对峰面积的RSD分别小于0.54%和2.08%,说明仪器精密度良好,符合含量测定要求[16]。(2)稳定性考察:按“2.1.2”制备S16供试品溶液,按“2.1.3”设定检测条件,分别于0、2、4、6、12、24 小时进样测定,以王百合苷C为参照峰,计算25个色谱峰与参照峰的相对保留时间及相对峰面积。计算结果显示,25个色谱峰与参照峰的相对保留时间和相对峰面积的RSD分别小于1.85%和2.68%,说明样品在24 小时内稳定[16]。(3)重复性考察:按“2.1.2”平行制备6份S16卷丹供试品溶液,按“2.1.3”设定检测条件并进样检测,以王百合苷C为参照峰,计算25个色谱峰与参照峰的相对保留时间及相对峰面积。计算结果显示,25个色谱峰与参照峰的相对保留时间和相对峰面积的RSD分别小于0.79%和2.13%,说明本方法重复性良好[16]。
2.1.5 代表性成分含量测定方法学考察 (1)线性关系考察:按“2.1.1”制备混合对照品溶液,按“2.1.3”设定检测条件并进样检测,得到各代表性成分峰面积,以对照品的量(μg)为横坐标,峰面积为纵坐标绘制标准曲线,计算回归方程及相关系数,结果见表2,线性方程相关系数r均大于0.999,表明线性范围符合测定要求[16]。
(2)精密度考察:按“2.1.2”制备S16供试品溶液,按“2.1.3”设定检测条件,连续进样6次,得到王百合苷C、王百合苷A、王百合苷E、王百合苷B及卷丹皂苷的峰面积,分别计算RSD为0.42%、0.36%、0.39%、0.31%和1.58%,说明仪器精密度良好,符合含量测定要求[16]。(3)稳定性考察:按“2.1.2”制备S16供试品溶液,按“2.1.3”设定检测条件,分别于0、2、4、6、12、24 小时进样,得到王百合苷C、王百合苷A、王百合苷E、王百合苷B及卷丹皂苷的峰面积,分别计算RSD值为0.70%、1.19%、1.16%、1.05%和1.71%,说明供试品溶液在24 小时内稳定性良好[16]。(4)重复性考察:按“2.1.2”平行制备6份S16卷丹供试品溶液,按“2.1.3”设定检测条件并进样检测,得到王百合苷C、王百合苷A、王百合苷E、王百合苷B及卷丹皂苷的峰面积并计算其含量,分别计算RSD值为0.38%、0.99%、1.38%、0.84%和1.69%,表明建立的含量测定方法重复性良好[16]。(5)加样回收率考察:精密称量6.03 mg卷丹皂苷对照品,用80%甲醇溶解并定容至2 mL容量瓶中(a)。精密称量15.10 mg王百合苷A对照品,用80%甲醇溶解并定容至2 mL容量瓶中(b)。取卷丹粉末约1 g,精密称定,各6份,分别加入a、b、王百合苷C、王百合苷E及王百合苷B对照品母液 0.28 mL、0.3 mL、0.1 mL、0.16 mL及0.25 mL,按照“2.1.2”项下方法,平行制备6份供试品溶液,按照“2.1.3”项下方法进样测定,得到王百合苷C、王百合苷A、王百合苷E、王百合苷B及卷丹皂苷的峰面积,计算王百合苷C、王百合苷A、王百合苷E、王百合苷B及卷丹皂苷的含量及加样回收率,结果见表3,回收率在95%~102%之间,RSD≤3%,表明建立的含量测定方法准确可靠[16]。
2.1.6 模拟基准点质量表征考察 精密称取王百合苷C对照品3.87 mg至50 mL容量瓶中,加80%甲醇溶解并稀释至刻度,摇匀,即得浓度为0.0774 mg/mL的王百合苷C对照品溶液作为模拟基准点,与“2.1.2”项下制备的卷丹饮片(S16)供试品溶液中所检测的王百合苷C浓度相同,按“2.1.3”设定检测条件并进样测定,得模拟基准点的HPLC色谱图,见图2。

表2 5个代表性成分的回归方程、相关系数和线性范围(n=5)
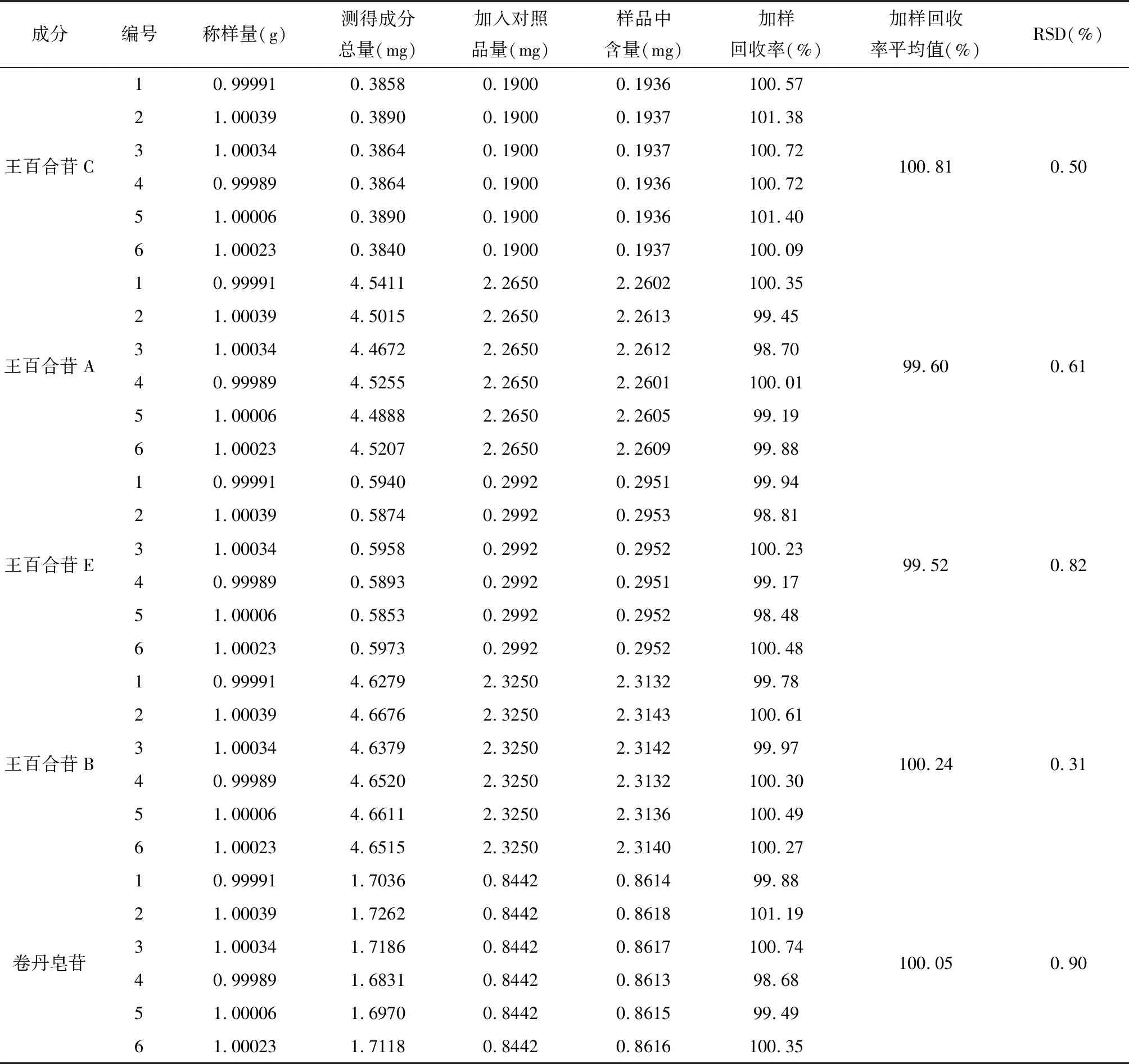
表3 加样回收率考察结果(n=6)
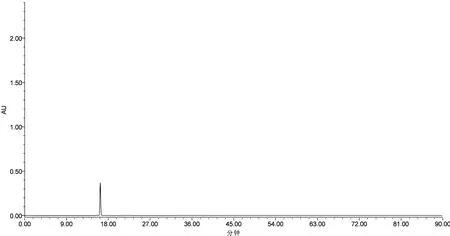
图2 模拟基准点的HPLC色谱图(330 nm)
由图2可知,王百合苷C对照品模拟基准点的保留时间(min)及峰面积(μV·s)与卷丹有效基准特征图谱中王百合苷C的保留时间及峰面积(有效基准饮片:16.200,4715466;模拟基准点:16.270,4747752)RSD值均小于3%,表明所建立的检测方法及其检测结果与模拟卷丹质量表征基准点均具有准确性,即王百合苷C对照品溶液(0.0774 mg/mL)可作为模拟表征点。
2.2 有效基准特征图谱质量表征信息
以S16为基准得到的有效基准特征图谱中26个特征峰的保留时间、峰面积(以100 mg卷丹饮片计)及含量,以模拟基准点为基准得到的相对保留时间、相对峰面积、相对含量及换算系数(代表性成分的相对含量/代表性成分的相对峰面积),见表4。
由表4可得卷丹有效质量基准标尺,即基于模拟基准点得到的26个特征峰及其相对保留时间、相对峰面积以及5个代表性成分的相对含量。
2.3 评价与验证
取15批待评价卷丹饮片样品,按“2.1.2”制备供试品溶液(n=3),按“2.1.3”设定检测条件并进样检测,得到15批待评价卷丹饮片质量表征信息及5个代表性成分的实测含量,同时实测S16卷丹饮片有效基准特征图谱,并通过模拟基准标尺得到5个代表性成分的换算含量,以验证基于有效质量标尺进行卷丹质量评价的准确性。如图3、4、5、6,以及表5所示。
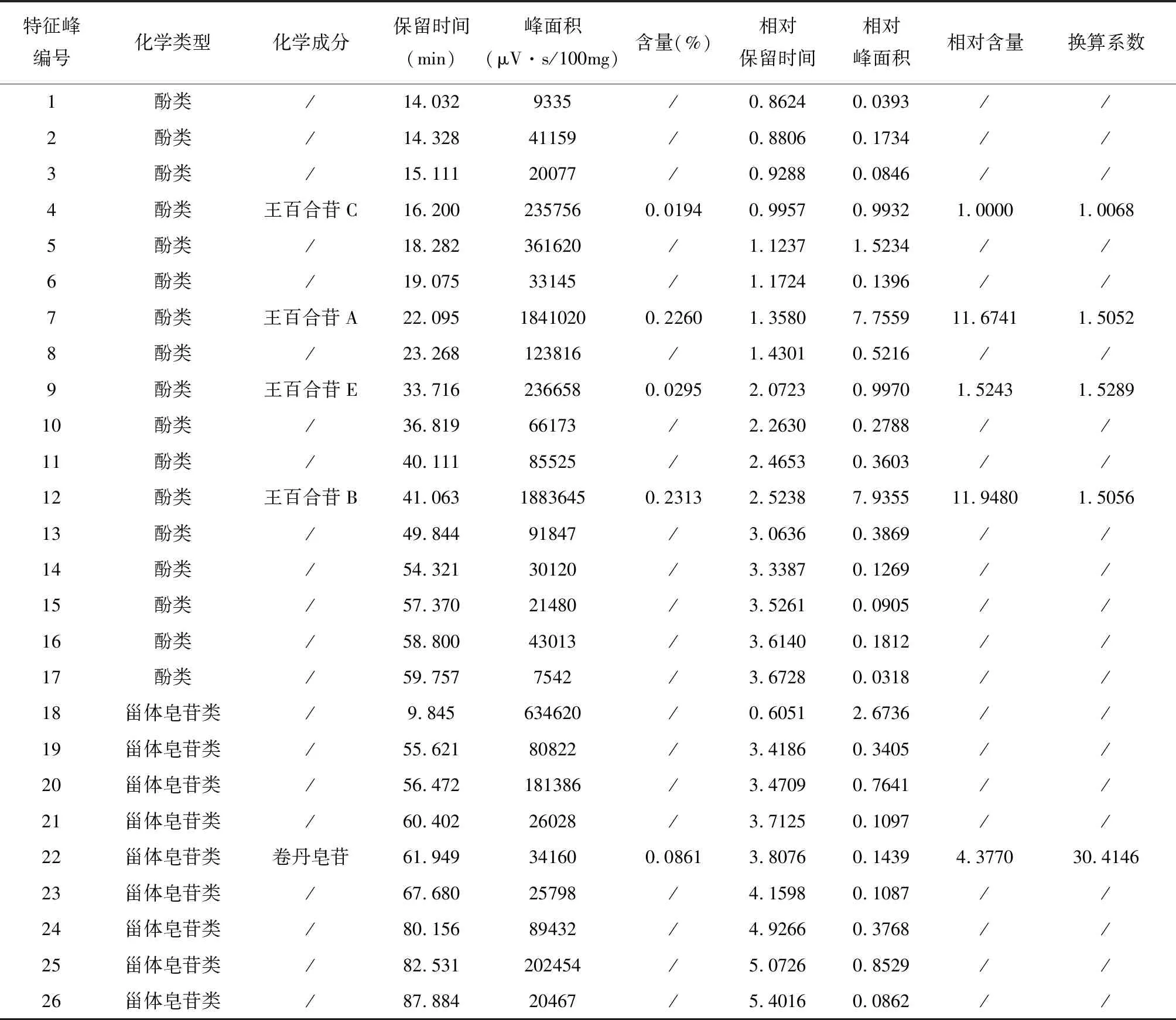
表4 26个特征峰的质量表征信息
注:A:330 nm;B:205 nm

表5 15批卷丹饮片中5个特征代表性成分的含量(%;n=3)
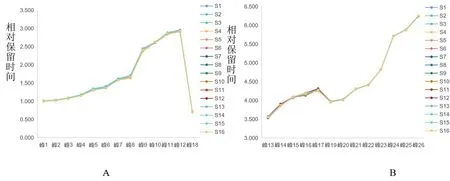
图4 15批卷丹饮片与基准(S16)的相对保留时间

图5 15批卷丹饮片与基准(S16)的相对峰面积

图6 15批卷丹饮片与基准(S16)的相对含量
如图3、4所示,15批卷丹饮片HPLC特征图谱皆含有26个相同的特征峰,即17个酚类特征峰及9个甾体皂苷类特征峰,且26个特征峰的相对保留时间与有效基准标尺(S16)基本一致。结果表明:15批待评价饮片皆为卷丹饮片,且说明了有效基准标尺可以应用于卷丹饮片真伪的鉴别。
由表5可得,15批卷丹饮片中5个代表性成分的实测含量与换算含量基本一致,即验证了代表性成分换算含量结果及其相对含量结果的准确性。
由图5、6可得,15批卷丹饮片质量表征信息,根据26个特征峰相对峰面积及5个代表性成分相对含量高于其有效基准标尺对应峰的数目和幅度,综合考量评价得出S7,S11,S10,S12,S5,S13和S4质量较优。
3 讨论
以自然药学观药物体系为导向[17-20],本研究基于HPLC-PDA技术,通过对供试品溶液制备方法(提取时间、提取溶剂、提取倍量及提取次数)及色谱条件(温度、流动相组成、流速及检测波长)进行考察,建立了卷丹有效基准特征图谱质量表征方法与评价模式。即以确定药效的饮片为基准,并以特征代表性成分对照品王百合苷C模拟特征图谱质量表征基准点,其特征图谱所表征的特征峰及其特征代表性成分质量信息涵盖卷丹主要化学类型酚类和甾体皂苷类,体现其质量内涵的整体性、有效性、基准性。本研究建立的卷丹质量评价方法能够整体、准确、便捷地对卷丹饮片进行质量表征,关注了卷丹饮片质量的精确性与应用有效性,为卷丹的质量评价与筛选及其药物质量的精准预期提供了科学依据。
