防污染PCR检测食源性沙门菌方法的建立
2021-03-16黄名英傅安静王远微
黄名英 , 傅安静 , 王远微
(1. 成都农业科技职业学院 , 四川 成都 611130 ; 2. 西南民族大学畜牧兽医学院, 四川 成都 610041)
沙门菌(Salmonella)是一种可导致人兽共患病的肠道菌科细菌,为两端钝圆的革兰阴性菌,兼性厌氧[1],无芽孢,一般无荚膜,除鸡白痢沙门菌和鸡伤寒沙门菌外,其他都有周身鞭毛。在世界食源性病原菌引起的食物中毒事件中,沙门菌经常位列榜首[2]。沙门菌在自然界中广泛存在,种类繁多,目前已检测出的沙门菌血清型有2 500余种,我国已报道的有292种血清型[3]。沙门菌营养要求低,繁殖快,在常温条件下即可繁殖,以肉类食品为主要污染对象,也可污染禽类、蛋类等食品[4-5]。传统方法检测沙门菌时间长,工作量大,特异性和灵敏度也不高[6],因此,如何快速准确检测出沙门菌对于食物中毒的防控极为重要。随着分子生物学技术的发展,沙门菌的检测方法也出现了聚合酶链式反应(Polymerase chain reaction,PCR)法[7-8]、实时荧光PCR法[9]、环介导等温扩增法(LAMP)[10]等。
PCR是一种用于放大扩增特定的DNA片段的分子生物学技术,它可看作是生物体外的特殊DNA复制,具有特异性强、灵敏度高、简便快速等特点,被广泛应用于食品检测中。但PCR技术过高的灵敏度导致其极易污染,产生假阳性结果,严重干扰PCR检测的准确性。而且随着技术不断发展,实时荧光PCR法和环介导等温扩增法等检测方法的灵敏度越来越高,造成污染的风险也越来越高。造成PCR污染的主要污染源是扩增产物和阳性对照物。由于拷贝数量极大,在PCR扩增时的开盖、摇动、吸样过程中与空气接触形成气溶胶体,对空气、试剂、移液器以及操作人员的手套衣物造成污染[11],反复造成阴性对照出现假阳性,导致检测无法正常顺利进行。为了防止PCR污染,很多技术方法被应用,主要有物理隔绝法、紫外照射法、水解法、化学修饰法和DEAE纤维素法等,但是这些方法费时费力,而且效果不理想。尿嘧啶糖基化酶(Uracil-N-glycosylase,UNG)是一种DNA修复酶,在适宜条件下能特异地识别并切割DNA分子中出现的尿嘧啶核苷。利用该酶的这种特性建立的防污染方法在一些病原检测中得到了应用,有效地消除了污染[12],但还未见该方法应用于食源性沙门菌的检测。
本试验基于UNG的生物学特性,用dUTP代替dTTP对沙门菌基因组DNA进行扩增,得到大量含有尿嘧啶的U-DNA作为检测阳性模板,在PCR反应体系中引入UNG和UNG抑制剂(UGI),对反应条件进行优化,最终建立防污染的沙门菌PCR检测方法。该方法对实验室环境要求以及操作人员的操作要求不严格,对于PCR反应的反应性、便捷性和时效性无影响,也不影响后续试验,同时可以解决阳性对照物以及扩增产物对PCR扩增体系污染的问题,降低假阳性的出现,提高检测方法的可靠性。同时,为建立基于UNG的防污染实时荧光PCR方法、LAMP方法等提供参考。
1 材料与方法
1.1 材料与试剂 鼠伤寒沙门菌CICC22956、单增李斯特杆菌CICC 21633,购自中国工业微生物菌种保藏管理中心(CICC);金黄色葡萄球菌、志贺氏大肠杆菌、空肠弯曲菌、链球菌、变形杆菌、绿脓杆菌等,均由西南民族大学动物医学实验室分离保存。UNG、TaKaRaTaqTM酶,均购自宝生物工程(大连)有限公司;UNG抑制剂(UGI),购自美国NEB公司;dNTP(dATP、dCTP、dGTP、dUTP),购自生工生物工程(上海)股份有限公司;细菌基因组DNA提取试剂盒,购自美国Sigma公司;营养肉汤、选择培养基,均购自杭州微生物试剂有限公司。
1.2 仪器与设备 PCR扩增仪,美国ABI公司;高速冷冻离心机,德国Eppendorf公司;电热恒温水浴箱,上海精宏实验设备有限公司;电泳仪、凝胶成像系统,美国Biorad公司;核酸定量仪,美国Thermo Scientific公司;恒温培养箱,美国 Thermo Scientific公司;微量移液器,德国Eppendorf公司。
1.3 方法
1.3.1 沙门菌的培养与DNA提取 把沙门菌菌株用平板划线法接种到营养肉汤上,在37 ℃恒温培养箱中培养24 h,然后取菌液接种到沙门菌选择培养基中,37 ℃下恒温培养24 h,传代培养2次,挑取单菌落扩繁,备用。
取沙门菌纯培养液经离心沉淀后,将得到的菌体沉淀按DNA提取试剂盒说明书提取细菌DNA,备用。
1.3.2 PCR方法的建立 根据GenBank中公布的沙门菌invA基因的序列,通过序列比对,找到保守序列,利用Primer Premier 7.0软件设计了1对引物。将设计的引物通过NCBI网站的BLAST工具进行检索,验证了其特异性。引物序列invA-F:ATTGGCGATAGCCTGGCGGTGG;invA-R:TCGCACCGTCAAAGGAACCGTA,扩增的目的片段大小为250 bp。引物由生工生物工程(上海)股份有限公司合成。
PCR反应按照TaKaRaTaqTM酶推荐的反应体系:DNA模板1 μL,上下游引物各1 μL(10 pmol),4×dNTPs(10 mmol/L) 2 μL,Taq酶(5 U/μL) 0.1 μL,Mg2+1.5 μL(25 mol/mL),10×PCR Buffer 2 μL,用超纯水补足至20 μL。
以上述PCR反应体系进行梯度PCR,优化退火温度,扩增程序:95 ℃预变性3 min;94 ℃ 30 s,50~60 ℃ 30 s,72 ℃ 30 s,30个循环;72 ℃温浴10 min。降至室温后使用2%琼脂糖凝胶电泳检测。
1.3.3 UNG防污染PCR反应体系的建立 将1.3.2中PCR反应体系中的dNTPs替换为用3倍浓度dUTP取代dTTP的dNTPs,同时相应的提高Mg2+的用量到2 μL,按照新的反应体系和优化好的退火温度,进行目的片段的PCR扩增。
1.3.4 UNG用量的优化 按照1.3.3所述反应体系和反应条件,1 μL的模板DNA的体系中分别加入1、1.5、2U和2.5U UNG,并设置无UNG对照,25 ℃下作用10 min,然后进行PCR反应,取5 μL产物进行2%琼脂糖凝胶电泳并在凝胶成像系统中观察分析电泳结果,确定模板完全降解时的最小酶用量,即为最适酶用量。
1.3.5 UNG反应时间的优化 按照1.3.3所述反应体系和反应条件,1 μL的模板DNA体系中根据1.3.4所得最适用量加入UNG,并设置无UNG对照,25 ℃下分别作用5、10、15 min和20 min,然后进行PCR反应,观察分析电泳结果,确定DNA模板完全降解时的最短反应时间。
1.3.6 UGI用量的优化 按照1.3.3所述反应体系和反应条件,1 μL的模板DNA体系中根据1.3.4所得加入UNG后,分别加入1、1.5、2U和2.5U酶抑制剂UGI,并设置无UNG对照和无UGI对照,先在50 ℃下作用20 min,再在25 ℃下作用1.3.5中优化的时间,然后进行PCR反应,观察分析产物电泳结果,确定UNG完全失去降解作用时的UGI用量,即为UGI最适用量。
1.3.7 UGI反应时间的优化 按照1.3.3所述反应体系和反应条件,根据1.3.4以及1.3.6所得,在1 μL模板DNA体系中加入UNG和UGI,并设置无UNG对照和无UGI对照,分别在50 ℃下作用5、10、15 min和20 min后,在25 ℃下作用1.3.5中优化的时间,然后进行PCR反应,观察分析电泳结果,确定UNG完全失去降解作用时的最短反应时间。
1.3.8 防污染效果验证 按照1.3.3所述反应条件,根据上述反应条件优化试验所得,在分别含有0.1、0.2、0.5、1 μg和2 μg阳性模板的PCR反应体系中,对照组不加UNG和UGI,A组中加入最适用量的UNG,B组加入最适用量的UNG和UGI,先在50 ℃条件下作用1.3.7中优化的时间,再在25 ℃条件下作用1.3.5中优化的时间,然后进行PCR扩增,观察电泳结果。
1.3.9 特异性检测 用优化好的防污染反应体系及反应条件分别对鼠伤寒沙门菌、单增李斯特杆菌、金黄色葡萄球菌、志贺氏大肠杆菌、链球菌、变形杆菌和绿脓杆菌样本进行检测,验证特异性。同时用未加UNG和UGI的反应体系和反应条件对上述样本进行检测,验证防污染体系对PCR方法特异性是否有影响。
1.3.10 敏感性检测 将已知浓度的阳性标准品进行10倍梯度稀释(101~1010)后作为模板,采用优化好的防污染PCR进行扩增,同时用未加UNG和UGI的PCR进行扩增,PCR产物用20 g/L琼脂糖凝胶电泳,检验灵敏度,同时验证防污染体系对PCR方法敏感性是否有影响。
2 结果
2.1 防污染PCR扩增体系和扩增条件 最终确定防污染PCR扩增体系:DNA 模板1 μL,上下游引物各1 μL(10 pmol),4×dNTPs(dUTP) 2 μL,Taq酶(5U/μL) 0.1 μL,Mg2+2 μL(25 mol/mL),10×PCR Buffer 2 μL,用超纯水补足至20 μL。扩增条件:95 ℃预变性3 min;94 ℃ 30 s,56 ℃ 30 s,72 ℃ 30 s,30个循环;再72 ℃温浴10 min。
2.2 含dUTP的目的DNA片段扩增 取沙门菌DNA作为模板,按照1.3.2中的方法配制成20 μL的反应体系进行PCR扩增,并对扩增产物进行2%琼脂糖凝胶电泳,观察结果,如图1所示,扩增后得到的产物片段大小为250 bp左右,条带清晰明亮。保留电泳结果为阳性的扩增产物为最后续试验的阳性模板。
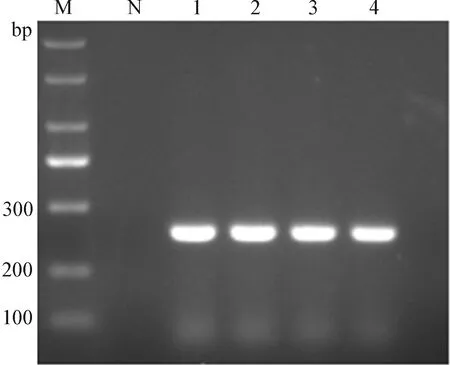
图1 目的DNA片段PCR扩增Fig.1 PCR amplification of target DNA fragmentM:Marker; N:阴性对照; 1~4:阳性模板M:Marker; N:Negative control; 1-4:Positive template
2.3 UNG用量的优化 取1 μL模板DNA进行UNG用量的优化,扩增后观察扩增产物琼脂糖凝胶电泳结果,如图2所示,经UNG作用后,扩增产物电泳条带变得模糊,清晰度变差,当UNG用量为1.5U时条带完全消失,即在25 ℃条件下作用10 min后,1.5U UNG可以将所有模板完全降解掉,UNG最适用量为1.5U。

图2 UNG用量的优化Fig.2 Optimization of UNG quantityM:Marker; 1:无UNG对照; 2:0.5U UNG; 3:1U UNG; 4:1.5U UNG; 5:2U UNG; 6:2.5U UNGM:Marker; 1:Without UNG control; 2:0.5U UNG; 3:1U UNG; 4:1.5U UNG; 5:2U UNG; 6:2.5U UNG
2.4 UNG反应时间的优化 在得出UNG最适用量的条件下,改变UNG作用时间,对UNG最适作用时间进行探索,结果如图3所示,随着作用时间的增加,UNG的降解能力增强,1 μg的模板DNA,加入1.5U UNG后,在25 ℃条件下作用15 min后,可以将所有模板完全降解掉,即UNG最适作用时间为15 min。

图3 UNG反应时间的优化Fig.3 Optimization of UNG reaction timeM:Marker; 1:无UNG酶对照; 2:5 min; 3:10 min; 4:15 min; 5:20 minM:Marker; 1:Without UNG control; 2:5 min; 3:10 min; 4:15 min; 5:20 min
2.5 UGI用量的优化 在得出UNG最适用量和最适反应时间的条件下,引入UGI,进行UGI用量的优化。结果如图4所示,UGI能够抑制UNG的作用,即随着UGI用量增加,UNG的降解作用减弱,条带的亮度增大。1 μg模板DNA,加入1.5U UNG和2U UGI后,先在50 ℃条件下作用20 min,然后再在25 ℃条件下作用15 min,可以完全使UNG失去降解作用。产物的电泳结果条带与无UNG时相同,即UGI最适用量为2U。
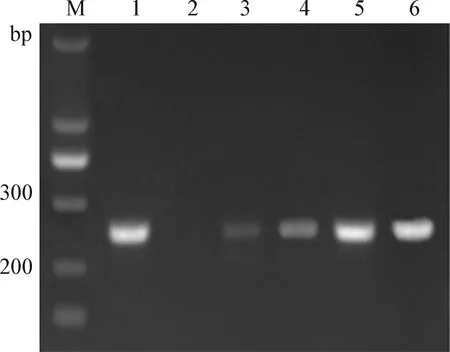
图4 UGI用量的优化Fig.4 Optimization of UGI quantityM:Marker; 1:无UNG对照; 2:无UGI对照; 3:1U UGI; 4:1.5U UGI; 5:2U UGI; 6:2.5U UGIM:Marker; 1:Without UNG control; 2:Without UGI control; 3:1U UGI; 4:1.5U UGI; 5:2U UGI; 6:2.5U UGI
2.6 UGI反应时间的优化 改变UGI反应时间,控制其他条件不变,对UGI反应时间进行优化。结果如图5所示,1 μg模板DNA,加入1.5U UNG和2U UGI后,先在50 ℃条件下作用10 min,然后再在25 ℃条件下作用15 min,可以完全使UNG失去降解作用,即UGI最适反应时间为15 min。
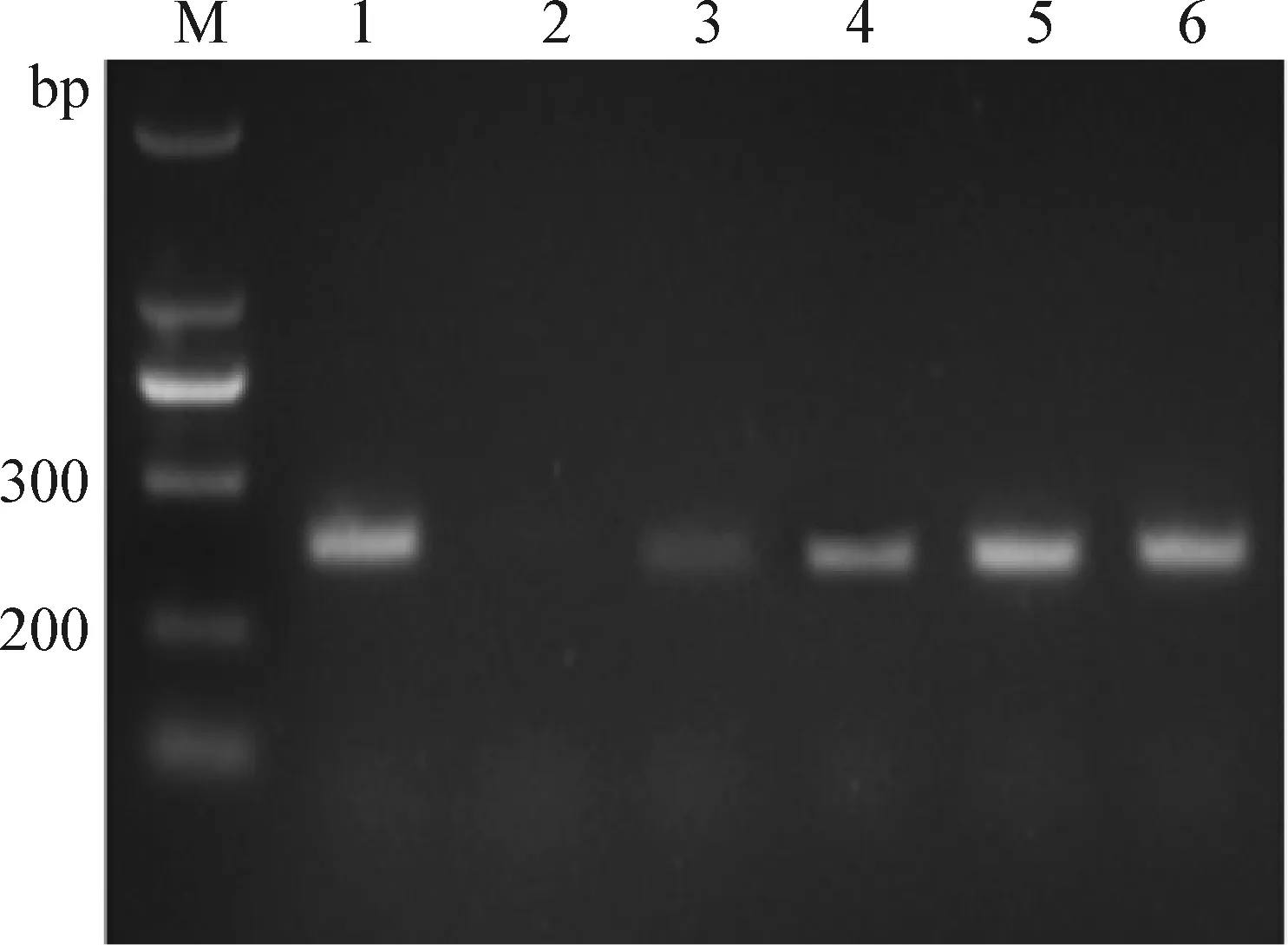
图5 UGI反应时间的优化Fig.5 Optimization of UGI reaction timeM:Marker; 1:无UNG对照; 2:无UGI对照; 3:5 min; 4:10 min; 5:15 min; 6:20 minM:Marker; 1:Without UNG control; 2:Without UGI control; 3:5 min; 4:10 min; 5:15 min; 6:20 min
2.7 结果验证 在含有0.1、0.2、0.5、1.0 μg和2.0 μg阳性模板的PCR反应体系中,对照组不加UNG和UGI,A组中加入1.5U UNG,B组加入1.5U UNG和2U UGI,先在50 ℃条件下作用10 min,再在25 ℃条件下作用15 min,然后进行PCR扩增,结果见表1,对照组所有浓度模板都有扩增,而且产物的量为“++++”,B组与对照组相同,A组在模板量为0.1、0.2 μg和0.5 μg的反应管为阴性,模板量为1.0 μg的反应管有微弱条带,条带亮度为“+”,模板量为2.0 μg的反应管条带亮度为“++”,表明UNG可以有效防止污染,虽然模板量为1.0 μg和2.0 μg的反应管有少量扩增,但是实际进行试验操作的时候很少能污染那么大量的阳性模板。UGI可以完全抑制UNG的作用,不影响产物的扩增。

表1 防污染效果验证结果Table 1 Verification of contaminant-preventing effects
2.8 特异性检测 特异性检测结果如图6所示,无论是防污染体系的PCR方法还是无UNG和UGI的PCR方法只能从鼠伤寒沙门菌阳性样本中检出相应的特异性条带,而对单增李斯特杆菌、金黄色葡萄球菌、志贺氏大肠杆菌、链球菌、变形杆菌、绿脓杆菌均没有扩增出条带,表明本试验所建立的PCR方法具有良好的特异性,而且加入UNG和UGI的防污染反应体系对PCR方法的特异性无影响。

图6 特异性检测Fig.6 Specificity testM:Marker; 1:鼠伤寒沙门菌; 2~7:分别为单增李斯特杆菌、金黄色葡萄球菌、志贺氏大肠杆菌、链球菌、变形杆菌和绿脓杆菌; 8:鼠伤寒沙门菌(无UNG和UGI的扩增体系); 9~14:分别为单增李斯特杆菌、金黄色葡萄球菌、志贺氏大肠杆菌、链球菌、变形杆菌和绿脓杆菌(无UNG和UGI的扩增体系)M:Marker; 1:Salmonella typhimurium; 2-7:Listeria monocytogenes,Staphylococcus aureus,Shiga’s Escherichia coli,Streptococcus,Proteus and Pseudomonas aeruginosa,respectively; 8:Salmonella typhimurium(Amplification system without UNG and UGI);9-14:Listeria monocytogenes,Staphylococcus aureus,Shiga’s Escherichia coli,Streptococcus,Proteus andPseudomonas aeruginosa,respectively (Amplification system without UNG and UGI)
2.9 敏感性检测 敏感性检测结果如图7所示,无论是防污染体系的PCR方法还是无UNG和UGI的PCR方法对鼠伤寒沙门菌的核酸最低检测限均为2.1×104copies/μL,具有较高的敏感度,而且加入UNG和UGI的防污染反应体系对PCR方法的敏感性无影响。

图7 敏感性检测Fig.7 Sensitivity testM:Marker; 1~6:分别为2.1×108 copies/μL至2.1×103 copies/μL的标准品; 8~12:分别为2.1×108 copies/μL至2.1×103 copies/μL的标准品(无UNG和UGI的扩增体系)M:Marker; 1-6:Standard substance from 2.1×108 copies/μL to 2.1×103 copies/μL,respectively; 8-12:Standard substance from 2.1×108 copies/μL to 2.1×103 copies/μL,respectively(Amplification system without UNG and UGI)
3 讨论
沙门菌是一种常见的人兽共患病原菌,在我国每年都会发生由沙门菌引起的食物中毒事件,如何快速准确地检测出沙门菌变得极其重要。PCR技术被广泛应用于沙门菌的检测中,但由于其自身特点而容易出现污染,为了解决这一难题,国内外的研究者提出了多种防污染技术,比如已经在目前的PCR试验中得到普遍应用的物理隔绝法、紫外照射法、水解法等。物理隔绝法主要通过对实验室操作平台进行严格分区,以防止试剂、样本及之间的交叉污染。紫外照射法操作较简单且成本低。近年来又发现了防污染效果更好的化学修饰法、DEAE纤维素法、酶消化法等。异补骨脂素法属于化学修饰法的一种,Cimino 等[13]将异补骨脂素在PCR前加入反应体系中,经过扩增后,再用UV照射15 min,结果发现异补骨脂素能够有效消除长度为500 bp以上的片段,但浓度100 mg/mL的异补骨脂素会降低PCR的效率[14]。Glushkov等[15]发现DEAE法最高能将污染降低106倍,但是此方法操作过程较为繁琐。
UNG法是酶消化法的一种,是利用UNG识别去除DNA中的尿嘧啶,把试剂中的dNTP全部替换为dUTP,扩增产物中的尿嘧啶被UNG消化后,在高温下断裂,而模板不含尿嘧啶所以不会受影响[16]。研究者在1974年发现UNG法,并纯化研究[17-19],至今被广泛应用于PCR防污染中。Longo等[16]验证了UNG法可以消除1010拷贝以上的 PCR 检测产物,且消化时间仅需10 min。Rios-Sarabia等[20]和Hwang等[21]在麻风分歧杆菌、HIV、莱姆病、衣原体PCR 检测中用此方法来防污染。Andersson等[22]将UNG处理与使用dUTP的预扩增相结合,为有效的消除污染、假阳性和不准确性提供了一种定量方法。Manajit等[23]采用尿嘧啶-DNA-糖基酶加环介导的恒温扩增技术,结合纳米金标记杂交探针(UDG-LAMP-AuNP)对铜绿假单胞菌进行检测,该方法快速、简便、特异,可用于铜绿假单胞菌污染样品的鉴定。Fallahi等[24]用dUTP取代dTTP成功进行了LAMP反应。在下一步反应前,用UNG选择性地裂解含有dUTP的LAMP产物,对含模板DNA无影响。这种改进的LAMP方法与UNG法处理相辅相成,可防止携带污染,为检测植物病毒提供了一种方法。
UNG法一方面可以有效地消除污染,另一方面对不含尿嘧啶的天然DNA片段不起作用,有效地保护了新生扩增产物,但是在反应体系中引入dUTP会降低PCR的扩增效率,因此需要对酶的用量、反应时间等条件进行优化[25]。本试验用3倍浓度dUTP取代dTTP,对沙门菌基因组DNA进行扩增,得到大量含尿嘧啶的U-DNA,将其作为后续试验的阳性模板。进行PCR反应前在反应体系中引入UNG,对UNG的用量和反应时间进行优化。UNG可将污染的含尿嘧啶的U-DNA水解产生脱嘌呤嘧啶位点,失去模板的功能,而在预变性过程中被灭活而不影响PCR扩增。在阳性对照组中加入UNG抑制剂来抑制UNG的活性,以保证阳性模板的顺利扩增,证明反应程序的正确性,并对抑制剂的用量和反应时间进行优化。最终建立的防污染方法为:在阳性对照的反应体系中加入1.5U UNG、2U UGI以及含有尿嘧啶的DNA(U-DNA)阳性模板;其他检测样品的反应体系中加入1.5U UNG,扩增体系所用的dNTP中以3倍浓度的dUTP代替dTTP,50 ℃保温10 min,再25 ℃保温15 min,然后再进行常规的PCR扩增,所有扩增产物均为含有尿嘧啶的U-DNA。试验验证结果表明,即使高浓度的U-DNA污染PCR反应体系,使用该方法也可以有效地防止污染,虽然模板量为1.0 μg和2.0 μg时仍有少量扩增,但是实际试验操作中很少出现这么大的污染量。特异性和敏感性检测结果显示,通过酶量和反应时间的优化,本试验建立的防污染PCR方法对于PCR的特异性和敏感性均无影响。
本试验建立的方法能有效快速地消除污染,操作简便,成本低,对实验室环境和操作人员要求不高,且对PCR扩增反应无影响,能够有效提高沙门菌PCR检测的准确性,防止假阳性的出现,为食源性沙门菌的检测提供了一种可靠的方法。
