硫化氢对糖尿病心肌病的保护作用
2021-03-06武韧常贵全孙凤起李鸿珠
武韧 常贵全 孙凤起 李鸿珠
(哈尔滨医科大学基础医学院/病理生理学教研室,黑龙江 哈尔滨 150086)
糖尿病心肌病(diabetic cardiomyopathy,DCM)是独立于高血压性心脏病和冠状动脉粥样硬化性心脏病的一种心血管疾病,是糖尿病常见并发症之一。DCM的主要病理学特征为心肌结构与功能的改变,包括心脏肥大、心肌细胞坏死、心肌间质纤维化和心肌内微血管病变等[1]。其发病机制尚未完全明确,可能涉及的机制包括:钙稳态失调、糖代谢与脂代谢紊乱、氧化应激损伤、炎症反应、肾素-血管紧张素-醛固酮系统失衡和细胞程序性死亡异常等[2-3]。硫化氢(hydrogen sulfide,H2S)是继一氧化氮和一氧化碳之后被发现的第三种气体信号分子,其在心血管疾病中的作用逐渐受到重视。目前已有研究表明H2S具有减轻心肌缺血再灌注损伤,抑制心肌细胞凋亡和减轻心肌氧化应激损伤等作用[4]。H2S对心脏的保护作用为DCM的研究提供了新的方向,现就H2S在DCM中的作用与机制展开综述。
1 H2S的生物学特性及作用
H2S是一种无色有臭鸡蛋气味的易燃气体,易溶于水。最初对H2S的研究主要针对其毒性作用,大剂量的H2S可抑制单胺氧化酶、细胞色素C氧化酶和碳酸酐酶等氧化酶,从而影响细胞能量代谢。后续研究发现H2S是一种内源性气体信号分子,广泛分布于机体各组织中,H2S的生理学作用成为研究的热点。人体内H2S的产生主要依赖胱硫醚-β-合成酶、胱硫醚-γ-裂解酶和3-巯基丙酮酸转硫酶三种通路,在心血管系统中以胱硫醚-γ-裂解酶通路为主[5]。生理浓度下的H2S对机体多种组织器官具有保护作用。在神经系统中,H2S可舒张脑血管,减轻神经胶质细胞诱导的炎症反应,调节神经内分泌功能,从而改善阿尔茨海默病和帕金森病的症状[6]。在呼吸系统中,H2S可减少活性氧(reactive oxide species,ROS)的生成,抑制肺动脉血管平滑肌细胞的增殖,减少细胞外基质的分泌和炎症因子的释放,减轻急性肺损伤和各种因素导致的肺血管重塑[7]。在泌尿系统中,H2S可减轻氧化应激损伤,抑制血管紧张素释放,扩张肾血管,减轻肾缺血再灌注损伤和肾脏纤维化[8]。在心血管系统中,H2S的作用更为广泛,H2S可激活血管平滑肌细胞钾通道蛋白,同时减少Ca2+内流,从而促进容量血管与阻力血管舒张,减轻心脏前后负荷,降低血压,改善高血压症状[9]。H2S还可通过抑制氧化应激和炎症反应保护受缺血再灌注损伤的心肌组织,减轻心肌间质纤维化[10]。在动脉粥样硬化方面,H2S可抑制单核细胞的黏附作用,减轻炎症反应,保护血管内皮细胞,抑制血管平滑肌细胞的增殖和迁移,从而减少动脉粥样硬化的形成[11]。此外,H2S可抑制高糖条件下心肌细胞凋亡和氧化应激并上调自噬,减轻DCM的心肌损伤[2]。
2 H2S对DCM的保护作用
2.1 H2S在DCM中的变化
有研究表明,与正常大鼠心肌相比,DCM大鼠心肌中H2S含量显著下降,补充外源性H2S的DCM大鼠左心室功能明显优于DCM大鼠,心脏肥大与纤维化也较轻[12]。在DCM小鼠的心肌与高糖培养的H9C2细胞中也可检测到H2S含量的下降[13],这说明内源性H2S的减少参与了DCM的病理生理过程,H2S对DCM的心肌具有保护作用。
2.2 H2S对DCM保护作用的机制
H2S可抑制DCM的发生,但其确切机制不详。现主要从H2S通过抑制氧化应激,抑制炎症反应,减少细胞凋亡和增加自噬,发挥减轻DCM的作用阐述H2S对DCM的保护作用(见图1)。
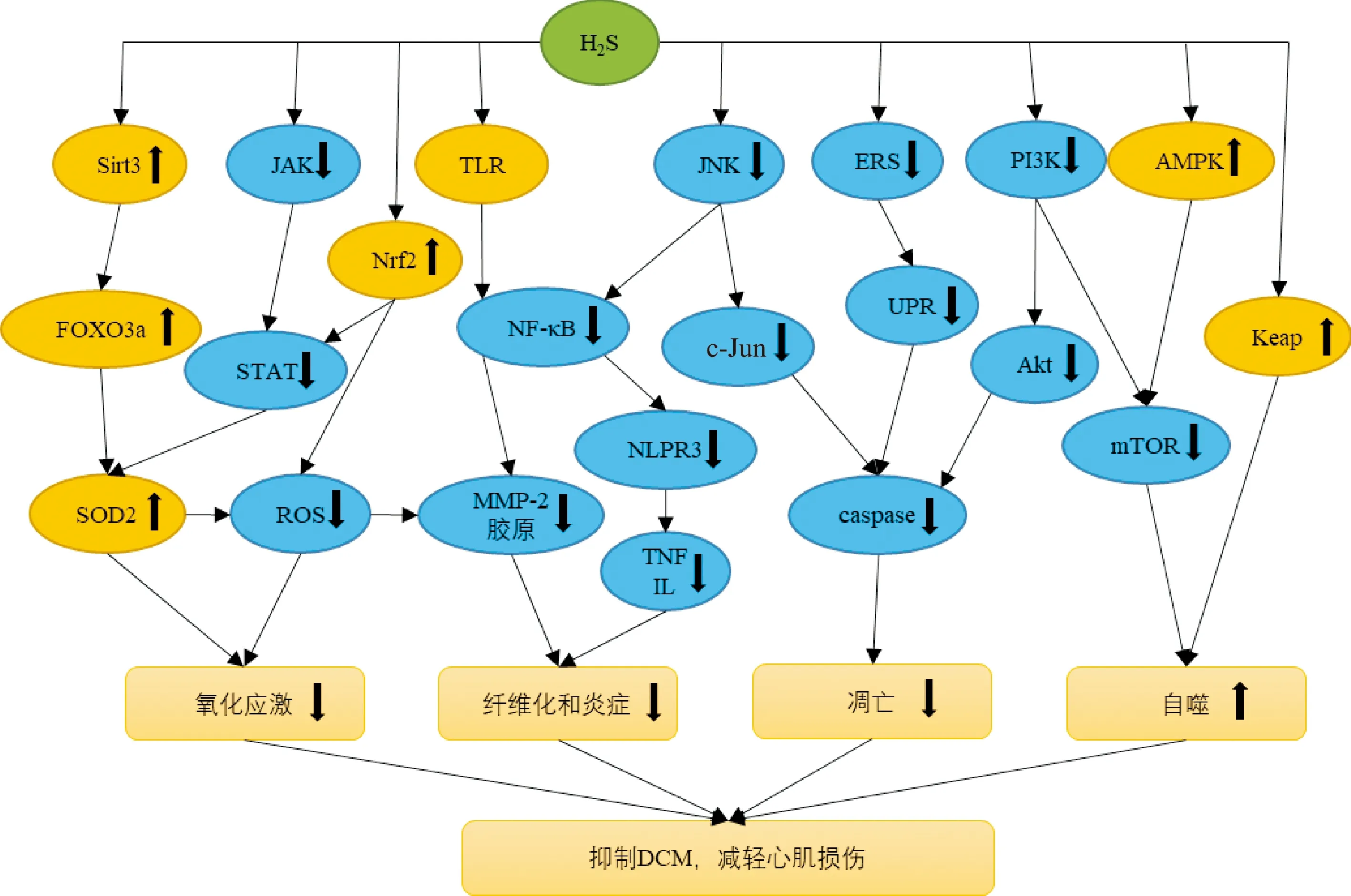
图1 H2S抑制DCM的机制
2.2.1 H2S与氧化应激损伤
氧化应激损伤是DCM发病的中心环节。DCM中,心肌细胞长期处于高糖环境,线粒体氧化代谢通量增加,电子传递链解偶联,ROS产生增多[14]。同时细胞内NADPH氧化酶、黄嘌呤氧化酶和脂氧合酶等氧化酶活性增加,细胞内氧自由基生成增多。此外,高血糖还可激活蛋白激酶C通路和晚期糖基化终末产物通路。过多氧自由基超出细胞清除能力,引发氧化应激,损伤DNA等生物大分子并激活细胞外调节蛋白激酶通路和c-Jun氨基端激酶(c-Jun N-terminal kinase,JNK)通路,细胞凋亡增加。激活的蛋白激酶C可进一步诱导核因子κB(nuclear factor-κB,NF-κB)的激活,引发炎症反应,进而导致心肌间质纤维化,心脏舒缩功能障碍。
作为一种还原性物质,H2S可直接通过HS-对ROS的直接淬灭产生抗氧化作用。间接作用方面,有研究表明,添加外源性H2S可快速诱导小鼠心肌细胞中核因子-E2-相关因子(nuclear factor-E2-related factor 2,Nrf2)的核转运,Nrf2可与抗氧化反应元件结合,增加血红素加氧酶-1等抗氧化蛋白的表达,减轻心肌氧化应激损伤[15]。H2S还可通过NAD依赖性去乙酰化酶-3通路上调FOXO3a和超氧化物歧化酶2的表达,从而保护线粒体功能,减轻氧化应激损伤[16]。也有研究发现在神经细胞中,外源性H2S可通过上调谷胱甘肽表达,降低氧化应激损伤,在心血管系统中也有一些实验发现了类似的现象。此外,H2S可通过多种方式减轻氧化应激损伤引发的心肌间质纤维化。H2S可下调NADPH氧化酶4的表达进而减少ROS,抑制细胞外调节蛋白激酶1/2通路,减少基质金属蛋白酶-2和胶原蛋白生成[17]。H2S还可通过Janus激酶-信号转导及转录激活蛋白通路减少细胞内ROS含量,抑制氧化应激损伤,减轻心肌间质纤维化[18]。
2.2.2 H2S与炎症反应
炎症反应的增加是DCM的重要发病机制之一。在DCM大鼠的心肌中,巨噬细胞数量显著增加,细胞间黏附分子-1、血管细胞黏附分子-1、白介素(IL)-1和IL-6等炎症因子含量上升[1]。大量炎症因子的释放可诱导细胞凋亡和心肌间质纤维化,影响心功能。DCM的心肌炎症反应与NF-κB通路的激活密切相关。高糖可通过多种途径激活NF-κB通路,NF-κB可激活NOD样受体家族3(NOD-like receptor-3,NLRP3)炎症小体,上调肿瘤坏死因子、IL-1和IL-6等炎症因子的表达,引发炎症反应。
H2S可通过抑制Toll样受体(TLR)-NF-κB-NLRP3通路减轻DCM的炎症反应。有研究表明高糖可诱导H9C2细胞TLR表达增加,TLR的激活触发信号级联反应,提高NF-κB活性,促进炎症因子释放。添加外源性H2S可降低NLRP3的表达,减轻炎症反应[19],有研究者认为这一作用通过翻译后修饰产生。H2S具有S-巯基化作用,可对特定靶蛋白的半胱氨酸残基进行化学修饰。有研究发现在人巨噬细胞中,H2S可通过S-巯基化作用修饰NF-κB的P65亚基,抑制NF-κB的磷酸化和核易位,从而抑制巨噬细胞的活化,减少炎症因子的释放。在心肌细胞中,H2S是否以同样的方式作用于NF-κB还有待研究。H2S还可通过ROS-JNK-NF-κB通路,减少ROS的产生,抑制JNK磷酸化,减少NF-κB核易位,减轻炎症反应[20]。NF-κB信号通路与DCM的心肌纤维化密切相关,有实验发现,链脲佐菌素诱导的糖尿病大鼠的左心室心肌中,NF-κB表达增加,转化生长因子-β1和基质金属蛋白酶-2含量上升,心肌纤维化明显。添加外源性H2S可增加细胞内谷胱甘肽含量,减少晚期糖基化终末产物生成,抑制NF-κB通路,减少转化生长因子-β1和基质金属蛋白酶-2的表达,减轻心肌纤维化[21]。此外,有研究发现,H2S可作用于冷诱导RNA结合蛋白-丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路,减轻高糖诱导的H9C2心肌细胞损伤和炎症[22]。
2.2.3 H2S与自噬
自噬是指从内质网脱落的双层膜包裹部分胞质和细胞内需降解的细胞器和蛋白质等成分形成自噬体,并与溶酶体结合,降解所包含物质的过程,具有实现细胞代谢需求和细胞器更新的作用。自噬在心血管疾病中的作用还未被完全阐明,一般认为适度的自噬水平可维持心血管内环境稳态,自噬的上调和下调都会引起细胞死亡,增加心血管不良事件风险[23]。自噬水平的下降与DCM的发生密切相关。有研究发现,糖尿病小鼠的自噬体数量减少,微管相关蛋白轻链-Ⅱ表达降低[24]。然而恢复自噬水平对DCM心肌的作用还存在争议。有实验表明,在提高糖尿病小鼠自噬水平后,心肌损伤反而较自噬水平降低组严重[24],也有实验得出相反的结果,白藜芦醇增加了糖尿病小鼠心肌细胞的自噬通量,减少了心肌损伤[25]。目前主流观点则认为恢复自噬水平对DCM心肌具有保护作用,也有一些研究者认为自噬水平的下降是DCM心肌细胞的自我保护机制。
H2S对自噬的作用在不同组织中存在差异,具有上调和下调自噬水平的双重作用。在DCM心肌中,H2S可调节自噬水平,保护心肌细胞功能,AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)-哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路是细胞自噬的重要途径。有研究表明,高糖培养的H9C2细胞中,AMPK磷酸化程度显著下降,其下游的mTOR活性上升,微管相关蛋白轻链-Ⅱ含量降低,自噬水平降低。外源性H2S可提高p-AMPK与AMPK比值,抑制mTOR,这说明H2S可通过AMPK-mTOR通路提高心肌细胞的自噬水平[26]。磷脂酰肌醇3激酶(phosphatidylinositide 3-kinase,PI3K)-蛋白激酶B(Akt)通路是细胞自噬调节的重要通路之一。H2S可提高糖尿病大鼠心肌细胞中PI3K-Akt-mTOR通路表达水平,抑制心肌细胞过度自噬,减轻心肌纤维化[27]。此外,H2S可通过抗泛素化作用提高细胞自噬。Kelch样环氧氯丙烷相关蛋白1(Kelch-like ECH-associated protein 1,Keap-1)可与抑制自噬的P62蛋白结合,参与自噬调控。H2S的S-巯基化作用可促进两个Keap-1分子间形成二硫键,阻止其泛素化,提高Keap-1活性,上调细胞自噬水平[28]。
2.2.4 H2S与凋亡
凋亡又称细胞程序性死亡,以胱天蛋白酶(caspase)诱导的蛋白水解级联反应为主要生化特征。凋亡机制复杂,已知有内在凋亡和外在凋亡两种主要途径。内在途径又称线粒体途径,由于不同原因导致的线粒体通透性改变,线粒体内细胞色素C等蛋白进入胞浆。细胞色素C与细胞凋亡蛋白酶激活因子1结合形成凋亡小体,激活下游caspase。外在途径又称死亡受体途径,肿瘤坏死因子-α等死亡配体与其受体结合,启动caspase-8和caspase-9,引发蛋白水解级联反应,大量研究表明高血糖可诱导心肌细胞凋亡。DCM心肌中炎症反应增加,大量的炎症因子可通过外在途径诱导凋亡,同时心肌细胞中氧化应激损伤可导致线粒体功能障碍,通过内在途径诱导凋亡。在DCM心肌中,糖毒性、钙失衡和自由基增多可导致内质网应激的产生。重度和慢性内质网应激可通过未折叠蛋白反应激活caspase-12依赖通路和C/EBP-同源蛋白依赖通路,诱导细胞凋亡[29]。心肌细胞凋亡造成细胞缺失,引发心脏舒缩功能下降和心脏重构。
H2S可通过多种途径减少DCM心肌细胞的凋亡。内在途径方面,H2S的抗氧化应激作用可保护线粒体功能,减少细胞色素C外漏,抑制凋亡。外在途径方面,H2S的抗炎作用可抑制NF-κB通路,减少炎症因子释放,减少死亡配体的含量,抑制凋亡。H2S还可通过减轻心肌细胞内质网应激抑制凋亡。有研究发现链脲佐菌素诱导的糖尿病大鼠心肌内质网应激的标志物葡萄糖调节蛋白78、caspase-12和C/EBP-同源蛋白含量上升,细胞凋亡增加。添加外源性H2S可下调葡萄糖调节蛋白78、caspase-12和C/EBP-同源蛋白的表达,从而保护心肌细胞[30]。此外,H2S可通过不同信号通路直接抑制凋亡相关蛋白的表达。有文献报道,H2S可作用于糖尿病大鼠心肌细胞MAPK通路和PI3K-Akt通路,降低P38-MAPK、JNK和Akt磷酸化水平,从而抑制凋亡相关蛋白的表达[12]。
3 小结与展望
H2S对DCM的保护作用已被大量实验证明。已有研究显示H2S可从减轻氧化应激,抑制炎症反应,上调细胞自噬和抑制凋亡等方面减轻DCM心肌损伤,保护心脏功能。然而H2S作为气体信号分子所具有的调节作用广泛而复杂,有很多在其他系统中得到验证的调节机制还未在DCM心肌中展开实验。H2S对DCM保护作用的机制依然具有很高的研究价值。
