20例糖原累积症Ⅸa型患者的临床和基因特点分析
2021-03-03李玉川冯佳燕王建设
李玉川, 陆 怡, 冯佳燕, 王建设
复旦大学附属儿科医院 a.肝病中心, b.病理科, 上海 201102
糖原累积症(glycogen storage disease, GSD)是一组由基因突变造成的参与糖原分解和糖原合成过程中酶缺陷导致的先天性糖代谢异常疾病,这些酶缺陷导致糖原在体内贮积,主要贮积在肝脏和肌肉。根据酶缺陷不同,GSD分为15型(0、Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅸ、Ⅹ、Ⅺ、Ⅻ、、、ⅩⅤ),其中Ⅰ、Ⅲ、Ⅳ、Ⅵ、Ⅸ型主要累及肝脏。GSD Ⅸ型比较常见,约占GSD的25%,该病是由于糖原磷酸化酶激酶(phosphorylase kinase, PHK)缺陷所导致[1]。根据致病基因的不同,GSD Ⅸ型可分为GSD Ⅸa、GSD Ⅸb、GSD Ⅸc和GSD Ⅸd 4型,其中GSD Ⅸa型最常见,该病是因PHKA2基因突变导致的肝脏PHK α亚单位缺陷,约占GSD Ⅸ型的75%[1]。GSD累及肝脏主要表现为肝肿大,转氨酶水平升高,空腹低血糖,高脂血症,生长发育迟缓。临床上,GSD Ⅸa型易被忽略,本文通过回顾性分析GSD Ⅸa型患者的临床资料,旨在总结GSD Ⅸa型的临床特征,加深对该病的认识。
1 资料与方法
1.1 研究对象 选取2015年1月—2018年12月于本院住院,并经临床及基因检查确诊为GSD Ⅸa型的患者。收集患者的临床资料,包括性别、年龄、身高、体质量、临床症状、体征、血常规、ALT、AST、肌酸激酶、空腹血糖、空腹血酮、甘油三酯、总胆固醇、乳酸、尿酮体、肝脏病理、基因结果、治疗及随访情况。
1.2 基因检测 收集患者的EDTA抗凝外周血2 ml,用QIAamp@DNA Mini Kit提取试剂盒(美国QIAGEN公司)提取全血基因组DNA。使用二代测序方法检测已知的20种GSD基因(GYS1、GYS2、G6PC、SLC37A4、GAA、AGL、GBE1、PYGM、PYGL、PFKM、PHKA2、PHKB、PHKG2、PHKA1、PGAM2、LDHA、ALDOA、ENO3、PGM1、GYG1)。测序结果采用Blast工具与美国国家生物技术信息中心(NCBI)提供的基因组序列进行比对,PHKA2使用的参考序列号为NM_000292。搜索千人基因组数据库和HGMD数据库,查看检测到的突变位点是否已有报道。用PolyPhen-2和Mutation Taster软件预测氨基酸替换对基因编码蛋白结构及功能的影响。
1.3 伦理学审查 本研究方案经由复旦大学附属儿科医院伦理委员会审批,批号:复儿伦审178号。所纳入患者均由其监护人签署知情同意书。
2 结果
2.1 临床资料 共纳入20例GSD Ⅸa型患者,均为男性,确诊年龄1.25~8岁,确诊中位年龄为2.5岁。所有患者均有肝肿大,且转氨酶水平升高。19例(95.0%)存在空腹低血糖,1例患者空腹血糖正常。身材矮小5例(25.0%);高乳酸血症14例(70.0%);高甘油三酯血症9例(45.0%);高胆固醇血症5例(25.0%)。8例患者进行了空腹血酮检查,结果均升高;5例(25.0%)患者尿酮体阳性;5例(25.0%)患者存在轻度贫血。20例患者均无肌无力、肌张力减低、运动发育迟缓;血常规白细胞、中性粒细胞、血小板均无降低,网织红细胞均无明显升高;肌酸激酶、尿酸水平均正常。纳入GSD Ⅸa型患者的临床资料详见表1。
2.2 肝脏病理检查结果 20例患者中有18例行超声引导下肝穿刺术,肝脏病理结果符合典型GSD表现,HE染色显示肝细胞肿胀,肝细胞弥漫性胞浆淡染,胞膜清晰,呈“植物细胞壁”样排列,胞质内见大量粉尘样物,PAS染色出现大量紫红色颗粒提示肝细胞糖原颗粒蓄积;部分患者MASSON染色显示不同程度纤维组织增生(图1)。肝脏病理纤维化程度S0 3例,S1~2 13例,S3 2例。余下2例患者拒绝肝穿刺活检,无相关病理结果(表1)。
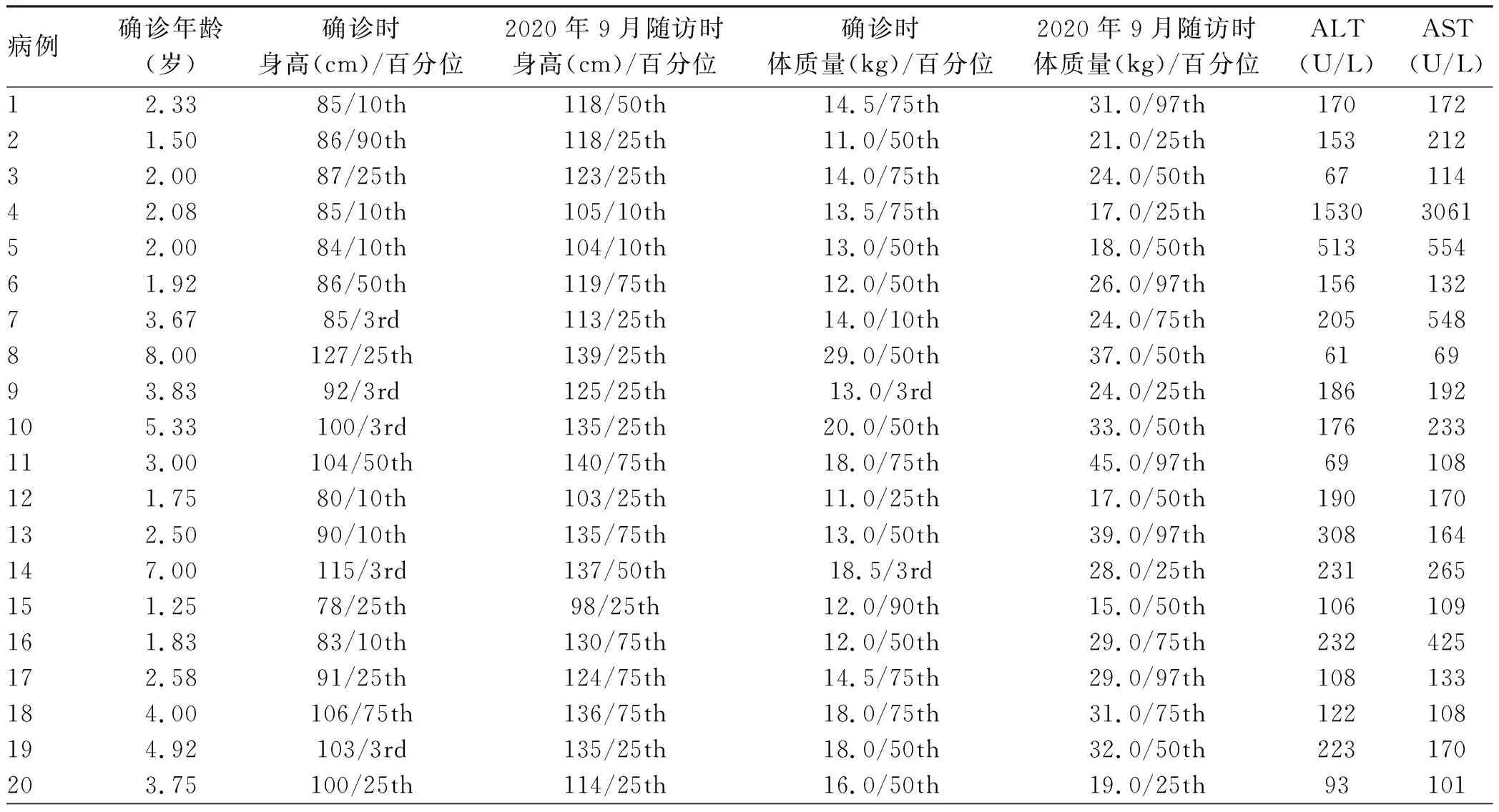
表1 20例GSD Ⅸa型患者的临床及病理资料
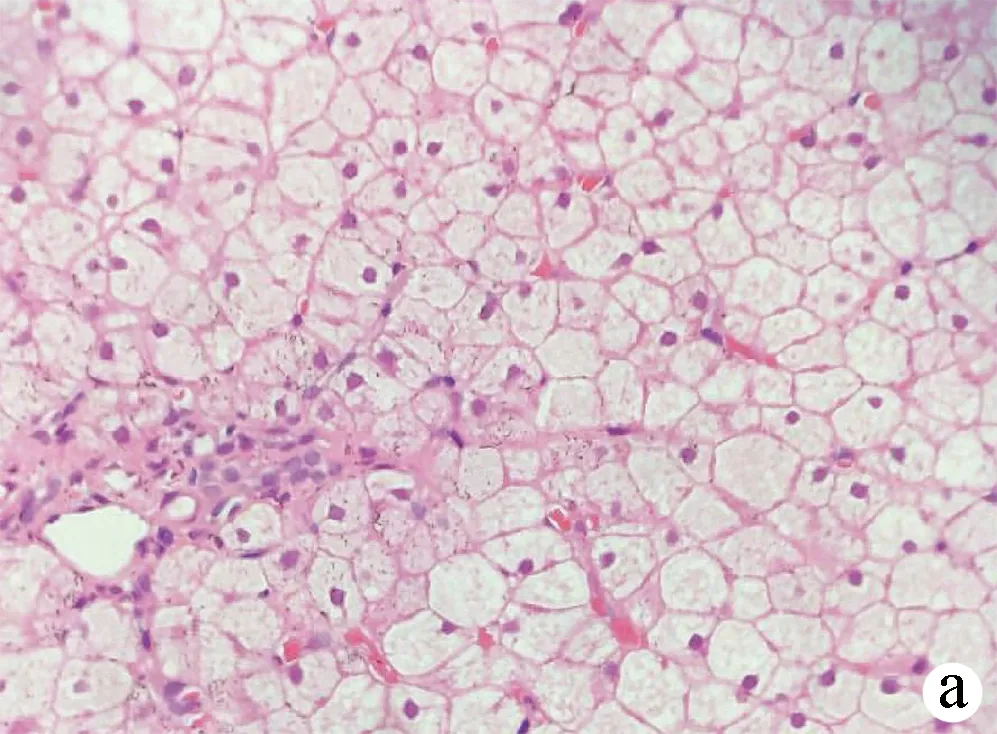
注:a,HE染色示肝细胞肿胀,肝细胞弥漫性胞浆淡染,胞膜清晰,呈“植物细胞壁”样排列(×400);b,PAS染色示糖原颗粒蓄积(×400);c,MASSON染色示汇管区纤维组织轻度增生(×400)。
2.3 基因检查结果 20例GSD Ⅸa型患者共发现16种PHKA2基因突变,均为半合子,包括10种错义突变、3种剪接突变、1种无义突变、1种缺失突变、1种小片段缺失。16种基因突变中有5种为已知致病突变,11种为新突变。新突变中包括6种错义突变,使用PolyPhen-2和Mutation Taster预测软件显示为致病突变;1种为无义突变,考虑为致病性突变;3种为剪接突变,考虑为致病性突变;1种为小片段缺失,考虑为致病性突变。有4例患者存在c.3614C>T(p.P1205L)基因突变,1例患者存在c.3614C>G(p.P1205R)基因突变;另有2例患者存在c.883C>T(p.R295C)基因突变。所有患者基因突变情况详见表2。

表2 20例GSD Ⅸa型患者PHKA2基因检测结果
2.4 治疗及随访情况 20例患者确诊后控制甜食的摄入,少量多餐高复合碳水化合物、足量蛋白质饮食,并给予生玉米淀粉口服治疗,每次0.6~2.0 g/kg,每6 h 1次。所有患者长期进行门诊随访。至2020年9月,随访1年9个月~5年8个月,门诊随访身高、体质量、肝脏触诊大小、血常规、肝功能、空腹血糖、肝脏超声。随访期间,17例患者规律饮食,口服生玉米淀粉,3例(病例2、4、5)规律饮食,未按要求规律口服生玉米淀粉。经治疗,20例患者空腹血糖均正常;17例患者肝功能转氨酶降至正常,3例(病例2、4、5) ALT水平波动在100~200 U/L,AST水平波动在120~200 U/L;19例患者肝脏触诊及腹部超声显示肝肿大明显好转,病例5仍有肝肿大,肝脏肋下10 cm,剑突下8 cm;18例患者身高、体质量增长,至2020年9月,身高百分位介于P25~P75,体重百分位介于P25~P97,5例身材矮小患者(身高3rd)身高增长(身高百分位介于P25~P50),病例4、5身高未见明显增长(P10)。
3 讨论
GSD Ⅸ型是由于PHK缺陷所致,导致不能激活糖原磷酸化酶,引起糖原分解障碍并在体内贮积,其发病率为1/10万[2]。PHK由α、β、γ和δ 4个亚单位组成,其具有广泛的组织分布,通过各种PHK相关基因表达和差异剪接产生多种组织特异性同工酶[3]。GSD Ⅸa型(OMIM*300798)是因PHKA2基因突变导致,PHKA2基因位于Xp22.2-22.1,包含33个外显子[4]。GSD Ⅸa型为X染色体隐性遗传病,男性发病率较高,本研究中20例GSD Ⅸa患者均为男性,符合其X连锁隐性遗传的特点。
文献[5-6]报道,90%以上的GSD Ⅸa型患者存在肝肿大和转氨酶水平升高,38%~48%的患者存在空腹低血糖。由于GSD Ⅸa型糖异生途径完整,减少了空腹低血糖的发生,因此,Ⅸa型患者严重空腹低血糖少见[7]。本研究中患者空腹血糖波动在2.0~4.5 mmol/L,平均3.1 mmol/L,与文献报道相符。由于低血糖时脂肪酸氧化代偿性增强,因此空腹血酮升高是GSD Ⅸa型的显著代谢特征[8]。本研究中8例患者进行了空腹血酮检查,结果显示血酮均升高,符合其代谢特征。14例(70.0%)患者乳酸升高,高于文献[6,9]报道。20例患者尿酸均正常,与文献[9-13]报道相符。临床上,GSD Ⅸa型、GSD Ⅰ型均可表现为肝肿大、转氨酶水平升高、高脂血症、生长发育迟缓,GSDⅠ型患者除了肝肿大,还表现为肾肿大、肾病,严重者可导致终末期肾病,由于血小板功能受损,GSDⅠ型表现为出血倾向,易出现鼻出血和瘀斑,贫血在GSDⅠ型患者中,特别是存在肝腺瘤的患者中比较常见,GSDⅠb 型患者临床上可出现中性粒细胞减少,易反复细菌感染[14-15]。本研究所有患者血常规提示白细胞、中性粒细胞、血小板均无降低,仅5例存在轻度贫血。此外,根据低血糖的严重程度及尿酸水平,也有助于二者之间的鉴别。有文献[5,7]报道GSD Ⅸa型患者可出现肌张力减低,运动发育迟缓,但肌酸激酶水平正常。本研究纳入患者均无肌无力、肌张力减低、运动发育迟缓,肌酸激酶水平均正常。综上GSD Ⅸa型患者的肝外表现有待进一步研究。
本研究中有18例患者行超声引导下肝穿刺术。肝脏病理检查有助于GSD的诊断,但具体分型仍需基因检查明确。18例患者肝脏病理显示15例存在不同程度肝纤维化,以轻度纤维化为主,13例(72.2%)患者纤维化程度为1~2级,2例(11.1%)患者纤维化程度为3级。虽然大多数GSD Ⅸa型临床表现较轻,预后较好,但也有进展为肝纤维化、肝硬化的相关报道[12,16-17],临床医生应提高警惕,注意密切随访,必要时行肝穿刺检查。
目前,HGMD数据库已收录151例患者,107种基因突变。包括7个剪接突变,9个插入突变,29个缺失突变,11个无义突变及51个错义突变[5],PHKA2以错义突变为主。目前在我国人群中已发现29种PHKA2基因突变[5-6,9,18-20]。本研究共发现16种PHKA2基因突变,以错义突变为主,其中5种为已知致病突变,11种为新突变。新突变的发现进一步丰富了PHKA2基因突变数据库。20例患者中4例存在c.3614C>T(p.P1205L)基因突变,1例患者存在c.3614C>G(p.P1205R)基因突变,提示c.3614位点是本研究的高发突变位点。中国、日本、韩国、丹麦、荷兰的人群中均有c.3614C>T的报道[1,11,21-24],在丹麦人群中发现该突变存在奠基者效应,亚洲或中国人群中是否也存在奠基者效应,尚不明确。韩国人群中,大多数突变类型为缺失突变或剪接突变,50%为大片段缺失,83.3%为剪接突变或缺失突变,韩国人群基因突变特点与其他种族不同[22,25]。本研究是国内纳入GSD Ⅸa型病例最多的一项研究,也是中国人群中PHKA2新突变最多的一项研究。本研究显示,我国人群PHKA2基因突变类型以错义突变为主,与国内其他学者的研究[6,9]相符。中国人群的PHKA2基因突变特点仍需进一步探究。
生玉米淀粉是治疗GSD的主要方法,生玉米淀粉具有分子量大、易在肠道停留的特点,口服后缓慢消化,逐渐释放葡萄糖,进而使血糖稳定在正常水平,减少肝脏的负荷。另外,生玉米淀粉还具有易获得、无特殊口味、价格便宜的特点,在临床上使用率较高。与GSDⅠ型相比,GSD Ⅸ型需要生玉米淀粉的量较小[7]。需要引起注意的是使用生玉米淀粉过度或不足均会对患者造成不利影响,过度治疗会导致糖原在肝脏贮积,还会引起腹泻、体质量增长过度、胰岛素抵抗,治疗的目标是使用适量的生玉米淀粉维持血糖在3.9~5.5 mmol/L,血酮在0~0.2 mmol/L[7,26]。本研究中20例患者在两餐之间使用生玉米淀粉口服,控制血糖稳定,控制甜食摄入,减少糖原产生,经治疗,大多数患者的生长发育水平,临床症状及肝功能、血糖好转,3例患者由于未规律口服玉米淀粉,生长发育落后,仍有肝肿大、转氨酶水平升高。国外报道[12]即使GSD Ⅸa型不治疗,患者也可达到正常的发育水平,转氨酶水平会随着代谢的改善和年龄的增长而下降,通常在成年期达到正常水平,肝肿大会在20岁后恢复正常[7,12,27]。虽然大多数患者预后较好,但也可导致肝纤维化,甚至肝硬化,因此,GSD Ⅸa型患者应规律随访。国外推荐,血生化、凝血功能应每3~12个月随访1次,18岁以下儿童应每12~24个月随访1次肝脏超声[7]。
综上所述,GSD Ⅸa型以男性为主,临床表现以肝肿大、转氨酶水平升高、生长发育迟缓、空腹低血糖、空腹血酮升高、尿酸正常为特征。GSD Ⅸa型肝脏病理多数表现为轻度肝纤维化,少数患者纤维化程度较重。本文报道了16种PHKA2基因突变,以错义突变为主,其中c.3614位点基因突变是高频突变位点。发现了11种PHKA2新突变,是国内单中心发现该基因新突变最多的一项研究,丰富了中国人群PHKA2基因突变图谱。GSD Ⅸa型大多数临床表现轻,早期干预,预后较好,长期规律随访,控制血糖稳定,改善代谢状态至关重要。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突,特此声明。
作者贡献声明:李玉川负责收集数据,资料分析,撰写论文;冯佳燕负责病理报告解读,提供病理图片;王建设参与课题设计;陆怡参与课题设计,拟定写作思路,指导撰写文章并最后定稿。
