A new mechanistic approach for the treatment of chronic neuropathic pain with nitrous oxide integrated from a systems biology narrative review
2021-03-03BaptisteBessireFranoisIrisAudeMiletAthanasiosBeopoulosCatherineBilloetraldineFarjot
Baptiste Bessière,François Iris,Aude Milet,Athanasios Beopoulos,Catherine Billoet,Géraldine Farjot,
1 Air Liquide Santé International,Paris Innovation Campus,Jouy-en-Josas,France
2 Bio-Modeling System,Paris,France
Abstract The limitations of the currently available treatments for chronic neuropathic pain highlight the need for safer and more effective alternatives.The authors carried out a focused review using a systems biology approach to integrate the complex mechanisms of nociception and neuropathic pain,and to decipher the effects of nitrous oxide (N2O) on those pathways,beyond the known effect of N2O on N-methyl-D-aspartate receptors.This review identified a number of potential mechanisms by which N2O could impact the processes involved in peripheral and central sensitization.In the ascending pathway,the effects of N2O include activating TWIK-related K+ channel 1 potassium channels on first-order neurons,blocking voltage-dependent calcium channels to attenuate neuronal excitability,attenuating postsynaptic glutamatergic receptor activation,and possibly blocking voltage-dependent sodium channels.In the descending pathway,N2O induces the release of endogenous opioid ligands and stimulates norepinephrine release.In addition,N2O may mediate epigenetic changes by inhibiting methionine synthase,a key enzyme involved in DNA and RNA methylation.This could explain why this short-acting analgesic has shown long-lasting anti-pain sensitization effects in animal models of chronic pain.These new hypotheses support the rationale for investigating N2O,either alone or in combination with other analgesics,for the management of chronic neuropathic pain.
Key words:central nervous system; chronic peripheric neuropathic pain; CPNP; integrative biology; laughing gas; medical gas; N2O;neuralgia; peripheral nervous system; receptors
INTRODUCTION
The World Health Organization has declared chronic pain,defined as any continuous or recurrent pain that lasts for more than 3 months,1to be a leading world health problem.2A subcategory of chronic pain is chronic neuropathic pain,which is chronic pain “caused by a lesion or disease of the somatosensory nervous system.”1In a large population survey from the USA,the 2017 prevalence of chronic neuropathic pain was estimated to be 15.7% among all reported pain(95% confidence intervals 14.9-16.5),3which is higher than the 7-8% prevalence of chronic pain with neuropathic characteristics reported by the International Association for the Study of Pain in 2014.4
The etiology of chronic neuropathic pain is varied,but common causes include diabetes,multiple sclerosis,shingles,spinal cord injury or spinal surgery,stroke,and trauma.1,5In case of neuropathies,pain arises spontaneously,can be elicited by normally innocuous stimuli (allodynia),is exaggerated and prolonged in response to noxious stimuli (hyperalgesia),and may spread beyond the site of injury (secondary hyperalgesia).1
The International Association for the Study of Pain recommends three types of treatment for first-line pharmacological management of chronic neuropathic pain:(1) secondary amine tricyclic antidepressants (TCA) or a dual serotonin noradrenaline reuptake inhibitor (SNRI); (2)calcium channel α2-δ ligands (e.g.,gabapentin,pregabalin);and (3) topical lidocaine for localized peripheral pain.6Opioid analgesics or tramadol are recommended first-line only for patients with acute neuropathic pain,neuropathic pain caused by malignancy,or for episodes of severe pain that occur as exacerbations of the chronic condition.6
This paper presents the results of a focused review using an analytical system biology approach to describe the biology of chronic neuropathic pain,taking into account the complex biological systems of pain signal transmission,pain perception,and central sensitization based on an integrative analysis of published data relevant to the mechanistic processes.With this understanding of the mechanical processes,a new theory has been formulated for the potential of nitrous oxide (N2O)to be used in this indication.Here we summarize the main findings of this approach,focusing on how N2O specifically may interact with the process of chronic neuropathic pain as a potential treatment.
METHODS
The authors carried out a review using a systems biology approach to integrate the complex mechanisms of nociception and neuropathic pain,and to decipher the effects of N2O on those pathways,beyond the known effect of N2O on N-methyl-D-aspartate receptors (NMDAR).The analytical procedure implemented (Computer-Assisted Deductive Integration) associates algorithmics and heuristics (Additional file 1).The logic behind this model-building approach does not assume functional linearity within biological systems and the components of a model do not incorporate solely what is known.Indeed,since this approach relies upon strict and systematic implementation of negative selection of hypotheses,models arising from this procedure contain elements that have never been described but cannot be refuted by current knowledge and/or available biological data,thereby generating novel understanding.This model-building approach has proven its efficacy in a number of biological research domains,including the discovery of hitherto unsuspected biological mechanisms,pathways,and interactions directly associated with phenotypic transitionsin vivo(be they pathological or developmental).7-15Further information on the Computer-Assisted Deductive Integration method can be found in a review from Iris et al.16
CATEGORIES AND MECHANISMS OF CHRONIC NEUROPATHIC PAIN
Neuropathic pain can be described as burning,stabbing,shooting,aching,or like an electric shock,but patients’ reports of pain vary in their description,character and intensity,all of which may be used to variables for classification.16However,such a classification rarely assists physicians in selecting the most appropriate therapy in chronic neuropathic pain conditions,especially when the underlying etiology of the pain (e.g.,diabetes,traumatic nerve lesion) cannot be cured.17Instead,classification based on the changes to the peripheral and central nervous systems that lead to the generation of abnormal pain sensitivity can provide mechanistic explanations for many of the temporal,spatial and threshold changes in pain sensibility in acute and chronic clinical pain settings.17Primary sensory neurons usually have a high threshold to stimulation so that only noxious stimuli will activate them.However,when tissue is damaged and these neurons are exposed to inflammatory mediators,their excitation threshold is lowered,increasing the responsiveness to nociceptive input.17This process is termed “peripheral sensitization” and it contributes to primary hyperalgesia at sites of inflammation.18Because the nociception changes occur at the site of tissue injury,ongoing peripheral pathology is required to maintain sensitization.19It has been shown that peripheral sensitization is implicated in the pathogenesis of altered temperature sensitivity,but not mechanical sensitivity.20Central sensitization produces pain hypersensitivity in noninflamed tissue by changing the sensory response elicited by normal inputs and increasing pain sensitivity long after the initial cause has disappeared or when no cause can be identified.21The molecular mechanisms responsible for central and peripheral sensitization are markedly different.21In central sensitization,neurons and circuits in nociceptive pathways throughout the central nervous system show increased membrane excitability,synaptic efficiency,or reduced inhibition.21These functional changes alter sensory inputs from the periphery causing the central nervous system to generate pain sensations in the absence of any particular peripheral stimuli.
KNOWN MECHANISMS OF NEUROPATHIC PAIN
Pain transmission pathway
The pain sensation occurs when a noxious mechanical,thermal or chemical stimulus is intense or prolonged enough to trigger an action potential in the usually resting peripheral nociceptors.22Nociceptor afferents have a unique structure,with both peripheral and central terminals utilizing the same biochemical processes,such that they can send and receive signals from either terminal.In peripheral tissues,the terminal nerve endings have a high threshold and do not transmit signals unless the stimulus presents a threat to physiological homeostasis.22Triggering the receptor sends an action potential through the afferent neuronal network,composed of unmyelinated C-fibers,and the myelinated Aδ and Aβ-fibers.22C-fibers respond to mechanical,thermal and chemical stimuli,but transmit signals more slowly than A-fibers,and are responsible for prolonged pain sensations such dull aches or a burning sensation.22Aδfibers are activated by noxious mechanical and thermal stimuli,and result in rapid high-frequency firing that is interpreted by the central nervous system as sharp localized pain.22Aβ-fibers usually carry input from innocuous stimuli like vibration,23but they can also respond to mechanical or thermal stimulation in the noxious range.22All of these neurons project to specific lamina of the dorsal horn of the spinal cord where they synapse with central projection neurons via second-order neurons.22
Nociceptive neurons release various excitatory neurotransmitters:C-fibers use neuropeptides (mainly substance P) and are therefore referred to as peptidergic,while glutamate is the key transmitter for Aδ-fibers.24Substance P interacts with G-coupled neurokinin-1 receptors on second-order neurons,activating a complex intracellular signaling cascade that results in nitric oxide production,and activation of both arachidonic acid and NMDAR pathways.24Glutamate,on the other hand,acts on second-order neurons via the ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor.24Other excitatory neurotransmitters in the dorsal horn include aspartate,vasoactive intestinal peptide,somatostatin,and calcitonin gene-related peptide.22
Within the dorsal horn,the incoming nociceptive signals are modulated by a complex network of central projection neurons descending from the brainstem,22and interneurons utilizing a range of endogenous molecules.The interneurons utilize mainly γ-aminobutyric acid (GABA) and glycine as inhibitory neurotransmitters,while the descending pain modulators use norepinephrine,serotonin,and opioid ligands.22,24,25Besides inhibiting excitatory neurons and terminals,noradrenergic and serotonergic fibers excite GABAergic and glycinergic interneurons.Norepinephrine depolarizes GABAergic neurons by activating α1-adrenoceptors (α1-AR),26while serotonin increases the frequency of GABAergic activity by activating 5-hydroxytryptamine 3 (5-HT3) receptors.27
Peripheral sensitization
As described earlier,peripheral sensitization may occur after nerve damage whereby action potentials in the normally highthreshold peripheral terminals are triggered at a lower stimulus threshold than usual.The increased electrical excitability of these neurons is mediated by upregulation in the expression of voltage-gated sodium channels.28Clusters of sodium channels accumulate not only at the site of the nerve damage,but also more centrally in the undamaged dorsal root ganglion.28This hyperactivity is exacerbated by downregulation of potassium channels such as TWIK-related K+channel 1 (TREK1),which would normally inhibit action potential propagation.29TREK1 is a potassium channel with a central role in anesthesia,neuroprotection,pain perception and depression.The complex gating properties of TREK1 are modulated by cell volume,actin cytoskeleton dynamics,cellular lipids,membranereceptor-coupled second messengers and pharmacological agents.30-33When TREK1 is downregulated,the ongoing afferent traffic initiated by the injury leads to the activation of spinal facilitatory cascades,yielding a greater output to the brain for any given input.
The sympathetic nervous system may also be involved in the generation of pain and other changes associated with pain states following trauma,ischemia,or deep-tissue microvascular vasomotor disturbances (complex regional pain syndrometype 1).Nerve injury triggers the expression of functional α1- and α2-ARs on cutaneous afferent fibers.In addition,the abnormal spontaneous activity at somatosensory nerve terminals triggers the sprouting of sympathetic neurons in the dorsal root ganglion.34 Postganglionic sympathetic neurons may activate receptors on the already sensitized primary afferent nerve terminals and cell bodies,either directly (on synapses via neurotransmitters such as norepinephrine and adenosine triphosphate) or indirectly (through secretion of norepinephrine into the blood); such activation may exacerbate the perception of the neuropathic pain.This magnifies the excitatory input in the dorsal root ganglion,34and is called sympathetically maintained pain.Sympathetically maintained pain is sensitive to treatments that target norepinephrine-mediated pain such as TCAs or clonidine.35
Central sensitization
Central sensitization develops when the central ascending nociceptive pathway becomes more excitable or there is a reduction in the inhibitory effects of the descending pathway.21Spontaneous activity within dorsal horn neurons develops or increases and the activation threshold of these neurons is reduced.21The result is that patients may experience pain in response to innocuous stimuli as well as noxious ones (allodynia),patients feel pain over a wider spatial area than that normally triggered by a stimulus (secondary hyperalgesia),or pain is persistent even after removal of the initial stimulus(sensation of “afterpain”).21
The development and maintenance of central sensitization is dependent on activation of glutamatergic receptors,and notably NMDARs.21Normally,NMDARs are maintained in a quiescent state by the presence of magnesium ions.With sustained nociceptive input,this usual voltage-dependent“brake” on Ca2+influx is removed,and these ions flow into the cell through the NMDAR pore.21This triggers intracellular signaling pathways including activation of Ca2+-dependent kinases,such as Ca2+/calmodulin-dependent protein kinase II,phosphatases (e.g.,calcineurin) and nitric oxide synthase.21In turn,these intracellular processes change the threshold and kinetics of the NMDARs and AMPA receptors,making the neurons more excitable,and promote expression of AMPA receptors on the synaptic surface.21Nitric oxide contributes to the increased excitability and reduces inhibitory activity within the dorsal horn neurons.21The disinhibited neurons become more susceptible to afferent inputs,including those from the normally non-nociceptive Aβ-fibers.21The C-fibers in the dorsal horn release the pain-promoting neuropeptide substance P that binds to neurokinin-1 receptor,36and accelerates the Ca2+influx through the NMDAR.Therefore,an efficient strategy to attenuate or block facilitation may be to reduce signal transduction,Ca2+currents and/or NMDAR activation.
CURRENT TREATMENTS FOR CHRONIC NEUROPATHIC PAIN
Because of the multiple receptors and neuromediators involved with the activation,amplification,and inhibition of nociception,analgesic agents can act at different sites within the pathway (Figure1).37Sympathetic blockers (e.g.,clonidine) block presynaptic α1-AR on afferent C-fibers,blocking the release of glutamate and substance P.24Anticonvulsants (carbamazepine,gabapentin,pregabalin,topiramate) have a range of effects including potentiating the activity of GABA,inhibiting sodium and calcium channels,and inhibiting NMDARs and AMPA receptors.38Dextromethorphan blocks NMDARs in the spinal cord.21TCAs,dual SNRIs inhibit serotonin and norepinephrine reuptake,enhancing their effect in endogenous pain-inhibitory systems in the brain.35Tramadol hydrochloride is a relatively weak central opioid analgesic,but it also acts as an inhibitor of serotonin and norepinephrine reuptake.6
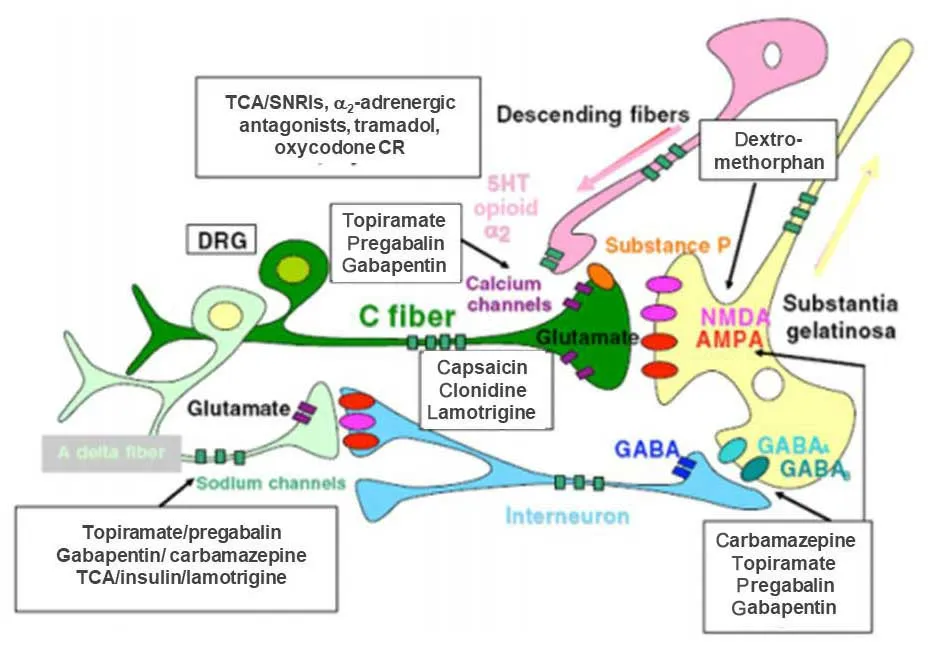
Figure1:The mechanisms of pharmacologic therapies for chronic neuropathic pain.
There is strong evidence for the use of antidepressants in clinical practice,in particular those that act on both norephinephrine and serotonin such as SNRIs and TCAs,for the treatment of neuropathic pain,including post-herpetic neuralgia,painful polyneuropathy,diabetic neuropathy,spinal cord injury,and multiple sclerosis.39-41However,they have not proven to be particularly effective in human immunodeficiency virus 1 neuropathy,phantom limb pain,or cancer related neuropathic pain.6Since chronic pain and depression often coexist,42the antidepressant effects of these agents may be an additional benefit.6
Gabapentin,pregabalin and carbamazepine are anticonvulsants indicated for the treatment of chronic neuropathic pain.Pregabalin is recommended by the National Institute for Health and Care Evidence as first-line treatment for patients with neuropathic pain.43However,the efficacy of these agents in neuropathic pain conditions is mixed.44-46
The use of opioids for chronic pain conditions of any etiology is increasing,47despite their known potential for adverse events such as paradoxical pain,abuse and addiction.48,49Because of inter-individual variability in response to opioids,50patients may receive higher-than-needed doses for the treatment of pain,increasing the risk of addiction,sleep apnea,respiratory depression and opioid-associated deaths.51,52Therefore,longterm opioid therapy is not suitable for most individuals with chronic neuropathic pain not related to cancer.53Opioids are only recommended as a second-line option when a combination of first-line agents is not optimally effective or feasible.6,53
The limitations of the currently available treatments for chronic neuropathic pain highlight the need for safer and more effective alternatives of chronic pain control.
NEW HYPOTHETICAL MECHANISMS OF N2O ON THE PAIN PATHWAY
N2O is traditionally used as a routine add-on to general anesthesia and for painful procedures.54Research in the last decade has found that N2O acts on one or more super-families of ligand-gated ion channels55causing multilevel blockade of the ascending and descending pathways,which effectively closes the gate to nociceptive inputs.As an NMDAR antagonist,56,57it produces moderate analgesia at sub-anesthetic concentrations and reduces surgical pain with a substantial neuropathic component.58N2O also facilitates the release of opioid peptides in the periaqueductal brainstem,activating the descending noradrenergic inhibitory pathways via GABA,thus reducing nociception at the spinal cord level.59
In addition to its acute effect,a single exposure facilitates post-injury pain rehabilitation and long-lastingly ameliorates severe neuropathic pain in rats,with the effect lasting for at least a month.60Therefore,the systems biology approach was used to identify possible rational mechanisms of action by which N2O may be able to interrupt the development of peripheral or central sensitization,and positively impact the course of chronic neuropathic pain.
Effects on the ascending pathway
In common with the inhalational anesthetic agents (chloroform,diethyl ether,halothane and isoflurane),N2O activates TREK1(potassium channel subfamily K member 2) in first-order neurons,leading to hyperpolarization of these presynaptic neurons and decreased release of glutamate (Figure2).61,62As described earlier,part of the peripheral sensitization mechanism involves downregulation of TREK1 channels,which dampens the pain inhibitory mechanisms in the dorsal horn lamina.30N2O may counteract this mechanism by activating TREK1.
N2O is also known to block CaV3.2 (T-type) voltagedependent calcium channels via free radicals.63This effect allows N2O to effectively attenuate neuronal excitability in nociceptive pathways,64while concomitantly reinforcing the effects of TREK1 opening by attenuating pre-synaptic glutamate release (Figure2).65
Another effect of N2O is to attenuate postsynaptic glutamatergic AMPA,kainate and NMDA receptor activation.55,56,66As described earlier,hyperpolarization of postsynaptic receptors removes the voltage-dependent Mg2+block of the NMDAR,causing increased neuronal excitability,but N2O antagonizes this hyperpolarization,thereby maintaining NMDARs in a non-activated state.65
Voltage-dependent sodium channels are responsible for the generation of action potentials and transmission in excitable cells.Blockade of sodium channels leads to the abolition of synaptic transmission and network function.67To date,there have been no reports of N2O acting on voltage-dependent sodium channels,but an inhibitory effect is likely,given that all major volatile general anesthetic agents (halothane,isoflurane,enflurane,ether and ethanol) have an antagonistic effect upon these channels.68N2O may affect serotonin-activated sodium/calcium channels by two mechanisms.First,N2O is reported to activate α2-ARs69-71while inhibiting β2-subunit-containing nicotinic acetylcholine channels,72which produces short-term inhibition of nociception mediated by muscarinic acetylcholine receptors,73,74and concurrently potentiating GABAAand glycine receptors.75,76
Second,N2O attenuates 5-HT3 receptor signaling,75,77thus promoting the postsynaptic inhibitory effects of GABAAreceptor activation while minimally affecting GABA and glycine release which is also positively controlled via presynaptic α2-ARs and muscarinic and nicotinic acetylcholine receptors.78-80
Two types of GABA receptors may be affected by N2O:GABAA receptors,which are ligand-gated chloride channels and thus responsible for fast inhibition of neurotransmission,81 and GABABreceptors,82which are G-protein coupled receptors that mainly influence potassium and calcium channels through second messenger systems,and result in a slower decrease of neurotransmission than that mediated by GABAAreceptors.82By potentiating GABAAand glycine receptors,76N2O appears to allow hyperpolarization (firing inhibition) of second order neurons that would otherwise favor depolarization,while concurrently favoring inhibitory depolarization in primary sensory neurons,thus attenuating nociception at two levels in the ascending pathway (Figure2).83
Effects of N2O on the descending pathway
Centrally,N2O exerts an analgesic effect by inducing the release of endogenous ligands of κ opioid receptors.83Activation of the opioid receptors by these endogenous ligands counteracts the tonic inhibition of descending noradrenergic pathways which is usually maintained by inhibitory GABAergic neurons.70,84In addition,by stimulating norepinephrine release,85N2O enhances the antinociceptive synergism between the α2-AR and the δ opioid receptor (DOR) (Figure2).86
Epigenetic effects
There is growing evidence that peripheral nerve injury causes epigenetic changes in pain-related pathways,and that these changes are implicated in the pathogenesis of chronic neuropathic pain.87,88By the same logic,inducing epigenetic changes may be a mechanism by which short-term treatment could induce long-lasting effects on pain regulation.
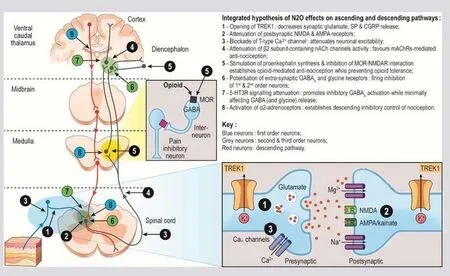
Figure2:Hypothetical mechanisms of action of nitrous oxide (N2O) in chronic neuropathic pain.
Epigenetic mechanisms regulate the transcription of ratelimiting metabolic enzymes,and are mediated by chemical modifications to chromatin,which modulate gene expression without changing the DNA sequence.89N2O causes the oxidation of vitamin B12 (cobalamine),preventing methyl transfer from the folate cycle,90and inhibiting the B12-dependent enzyme methionine synthase.89This causes a depletion of S-adenosylmethionine (Figure3).88S-adenosylmethionine is the main cofactor for DNA methylation now considered to be a key mechanism for regulating gene expression in neurons.91
N2O may mediate epigenetic changes via the opioid pathway.For example,when S-adenosylmethionine is downregulated,the downstream transcriptional effect is to increase production of δ- and κ-opioid ligands (principally met-enkephalin) at the expense of µ-receptor peptides (β-endorphin) and nociceptin.92A reduction in DNA methylation may also increase the expression of DOR and μ-opioid receptors (MOR),but does not affect κ opioid receptor expression,which is regulated by histone acetylation.93
N2O-induced epigenetic regulation of µ-peptides and MOR expression should decrease the NMDAR-MOR association and sensitization induced by NMDAR activation.94This is reminiscent of the inhibition of morphine tolerance by administration of NMDAR antagonists,such as ketamine or dextromethorphan.93Apart from breaking the opioid tolerogenic MOR-NMDAR interaction,an epigenetically mediated increase in the ratio of DOR ligand expression (e.g.,metenkephalin) would also favour a DOR-α2AR interaction,95which may promote long-lasting anti-nociceptive synergistic effects of N2O.
DISCUSSION
The results of the current review provide a rational explanation for a potential therapeutic benefit of N2O in patients with chronic neuropathic pain,and this is supported by data from animal studies suggesting that N2O has short- and long-lasting analgesic effects.96Usage of N2O in acute analgesia is well established in clinical practice,however its secondary effects linked to B12 inactivation prevents a daily repeated usage.The long lasting analgesic effect,may overcome this shortage but needs to be proven in clinical trials on established chronic neuropathic pain.However,to date,only one clinical study with N2O has been undertaken in patients with chronic neuropathic-type low back pain,and this study did not show a significant benefit compared with oxygen in a small cohort of patients (n= 70).97One possible explanation is linked to the different etiology of low back pain that has an important inflammatory component,being released by anti-inflammatory medications,contrary to chronic neuropathic pain.

Figure3:Hypothetical link between oxidation of vitamin B12 by N2O and epigenetic regulation as a basis for long-lasting effects on pain regulation.
As with many other pain conditions,multimodal therapy is often required for patients with chronic neuropathic pain,and N2O may have a role as adjunctive therapy,enhancing the analgesic effects of other agents.N2O mediates activation of the anti-nociceptive descending pathway,together with attenuation of MOR-NMDAR interaction and concurrent potentiation of extrasynaptic GABAA and glycine receptors,α2-ARs,noradrenaline release,pro-enkephalin production and DOR-α2AR interaction.68,83Therefore,pre- or co-medication with N2O could allow a reduction in the therapeutic doses of other analgesic agents,limiting the occurrence of undesirable adverse events,without compromising therapeutic efficacy.For example,N2O uncouples the MOR-NMDAR interaction,which would help to re-sensitize MORs and limit opioid tolerance.By activating the α2-ARs,N2O might allow for a lower dose of opioid to be used,while the attenuating effects of N2O on 5-HT3 signaling might also facilitate a reduced dose of SNRIs.
N2O might also have a role as an adjunct to anticonvulsant therapy for the treatment of chronic neuropathic pain.Anticonvulsants often need to be used in high doses to achieve analgesic effects in chronic neuropathic pain,placing patients at risk of unwanted adverse events.98Since both N2O and anticonvulsants potentiate GABAergic nociceptive pathways,using the two in combination might allow a lower dose of anticonvulsant to be used.However,caution should be exercised since the negative effects of N2O on vitamin B12-dependent mechanisms of erythropoiesis could increase the risk of hematologic adverse events with some anticonvulsants such as carbamazepine.99,100
SUMMARY AND CONCLUSIONS
Chronic pain is a leading health problem,affecting a large number of people worldwide.2Moreover,the pathophysiological processes of chronic pain development are complex,and reflect the plasticity of the peripheral and central nervous systems.36Because no single mechanism is responsible for chronic pain,treatment is also complex and usually involves multimodal therapy targeting more than one nociceptive pathway.Chronic pain management requires a thorough assessment of a patient’s pain condition as well as any underlying causes.6Current pharmacologic management strategies for chronic pain are often limited in maintaining long-term pain control and are all associated with potential issues,such as adverse events,dependence or tolerance.
The current algorithmic and heuristic analysis was designed to identify a potential interface between the mechanism of chronic pain and the pharmacological effects of N2O in areas of the brain and spinal cord where chronic pain originates.The findings suggest that there are supportive reasons for N2O to have beneficial effects in the treatment of chronic neuropathic pain,not only via targeting NMDA receptor.Ongoing research is exploring the clinical potential for N2O in this setting.
Acknowledgements
We would like to thank Catherine Rees and Mimi Chan,PhD,of Springer Healthcare Communications,for medical writing assistance.We thank Ira Katz,of Air Liquide Santé International,for critical proof reading.
Author contributions
GF conceived the project and proof outline.BB and AM brought their expertise in preclinical neuroscience and pain.CB ensured the medical reliability of the content with her experience of clinical pain treatment.GF,BB,AM,CB equally contributed to the final version of the manuscript.FI and AB performed the systems biology computation and elaborated the mechanistic theories.All authors approved the final version of the manuscript for publication.
Conflicts of interest
BB,AM,CB and GF are employees of Air Liquide Santé International(ALSI).ALSI is currently marketing N2O in analgesia,as an add-on to general anesthetic agents and for the prevention of procedural pain in a fixed 50% mixture with O2.ALSI is also conducting a clinical trial (EudraCT #:2015-004779-64) with N2O in chronic neuropathic pain.FI and AB are employees of Bio-Modeling Systems,funded by Air Liquide Santé International for this system biology analysis.
Financial support
This work was supported by Air Liquide Santé International.
Copyright license agreement
The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check
Checked twice by iThenticate.
Peer review
Externally peer reviewed.
Open access statement
This is an open access journal,and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional file
Additional file 1:Computer-Assisted Deductive Integration method.
杂志排行

Medical Gas Research的其它文章
- High-flow hydrogen inhalation might suppresses the immune function of middle-aged participants:a selfcontrolled study
- The effect of dexmedetomidine on decrease of cough,hemodynamic parameters and Ramsay score versus lidocaine during general anesthesia:a randomized clinical trial
- Evaluation of audible leak versus pressure volume loop closure for polyvinyl chloride cuff and polyurethane microcuff in endotracheal tube inflated with air:a prospective randomized study
- The role of nitric oxide in peptic ulcer:a narrative review
- Therapeutic effects of hyperbaric oxygen:integrated review
- Hyperbaric oxygen therapy decreases mortality due to Fournier’s gangrene:a retrospective comparative study