根癌农杆菌vbp2基因启动子转录调控的探析
2021-02-10徐楠徐宇娟孙盼宗仁杰郭敏亮
徐楠 徐宇娟 孙盼 宗仁杰 郭敏亮
(扬州大学生物科学与技术学院,扬州 225009)
根癌农杆菌(Agrobacterium tumefaciens),是一种广泛存在于土壤中的革兰氏阴性菌。作为一种常见的植物病原菌,它能够使大多数双子叶植物发生冠瘿瘤疾病。其致病机制是将自身Ti质粒上的部分DNA片段(简称T-DNA)以T-复合物的形式转运到宿主植物细胞,并整合到宿主的基因组中,使宿主植物细胞发生遗传转化而致病[1-3]。由于根癌农杆菌能将基因转给宿主植物,因此,根癌农杆菌很快就成为了应用最广泛的植物转基因工具菌[1-6]。根癌农杆菌的致病(或转基因)受宿主分泌的糖类、酚类等小分子化合物的诱导[7-9]。当环境中没有诱导物时,绝大多数与致病有关的基因都不表达,根癌农杆菌不具备致病能力[9]。当诱导物存在时,就可以诱导相关的vir基因表达出一系列致毒蛋白(virulence protein),其中的VirD2蛋白具有核酸内切酶活性,从Ti质粒上切下单链的T-DNA,并通过共价键与T-DNA的5′-端连接形成VirD2-T-DNA复合物,进而加工成T-复合物[1-3,10-12]。T-复合物通过根癌农杆菌细胞两端的四型分泌系统(type IV secretion system,T4SS)进入植物细胞,其中VirC1蛋白和VBP(VirD2-binding protein)蛋白可以提高T4SS对T-复合物的招募作用[13-15]。
VBP蛋白,又叫T-复合物招募蛋白,是一种可以与VirD2专一性结合,并参与T-DNA转运的蛋白[14]。其具体功能是将T-复合物招募到根癌农杆菌细胞膜上的T4SS处,促进T-DNA的转运。根癌农杆菌基因组中含有3个能编码VBP的平行同源基因,分别为vbp1(atu5117)、vbp2(atu4860)、vbp3(atu4856)[13-14]。只有3个vbp基因全部敲除,根癌农杆菌的致瘤能力才大幅降低[13],3个同源基因在招募T-复合物上具有功能互补性。但是,从进化的观点看,相对精简的原核基因组为一项非必需功能保存3个同源基因,有些不太经济,因此,我们推出VBP蛋白可能还具有招募T-复合物之外的其它功能[14]。为了进一步探寻VBP的其它功能,我们对3个vbp基因在致瘤诱导条件下的表达情况进行了研究,发现vbp1和vbp3基因的表达不受致瘤诱导条件的影响,而vbp2基因的表达却受致瘤诱导条件的影响,同时还发现vbp1的缺失也会影响vbp2的表达[16]。这些研究表明3个vbp基因也可能相互之间影响彼此的表达。进一步研究vbp2基因转录表达调控的机制,不仅对优化根癌农杆菌转基因的诱导条件,提高转基因效率具有实践指导意义,而且对深入解析VBP蛋白的其它生物学功能也具有重要的参考价值。
本研究在生物信息分析vbp2启动子结构的基础上,通过构建原位替代突变株,确定了不同诱导条件对vbp2启动子的调控作用,以及最适的诱导浓度。根据生物信息学分析预测,通过启动子截短的方法,确定出诱导作用调控元件所在的区域。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 根癌农杆菌(A. tumefaciens)C58为胭脂碱型野生菌,vbp1和vbp2缺失的根癌农杆菌菌株为 GMI9017Δvbp2,由 C58 衍生而来[13],大肠杆菌宿主菌为 DH5α(Escherichia coli DH5α);自杀质粒pEX18Km、表达载体pUCA19、pCMV6-AN-RFP(红色荧光蛋白基因的质粒),eGFP-T(绿色荧光蛋白基因的质粒)均为本实验室保存。本文所用到的引物如表1所示。
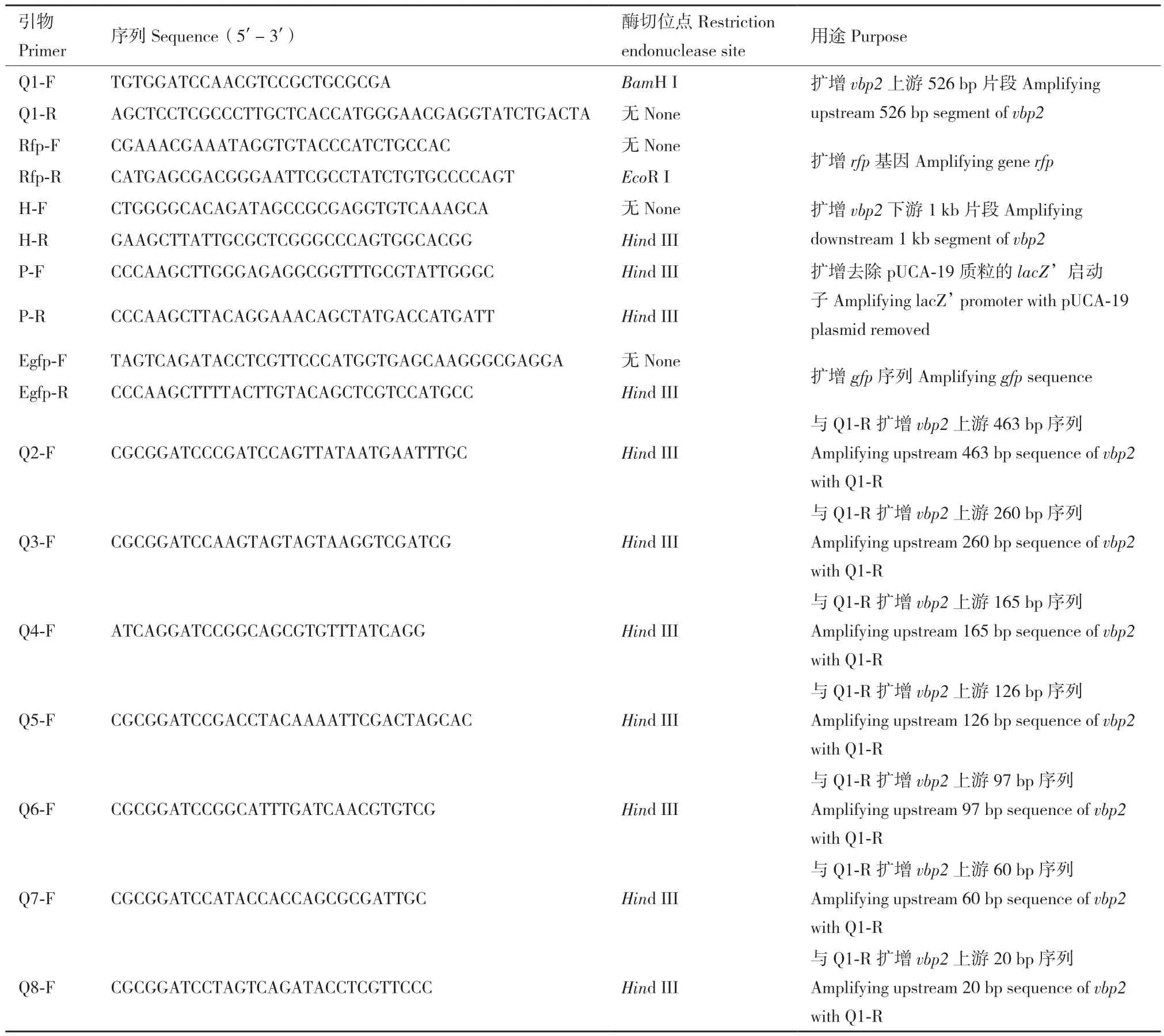
表1 本研究所使用的引物Table 1 Primers used in this study
1.1.2 培养基 大肠杆菌用LB培养基培养,根癌农杆菌用MG/L和AB蔗糖培养基培养,具体配方见文献[13-14,16]。
1.2 方法
1.2.1 vbp2启动子预测和分析 在HMM Database(www.ebi.ac.uk/Tools/hmmer/.)、Prokaryote Promoter Prediction(https://services.healthtech.dtu.dk/.) 和TESS(http://agave.humgen.upenn.edu/utess/tess) 网站对A. tumefaciens农杆菌T-复合物招募蛋白VBP2的编码基因vbp2上游序列中可能存在的转录因子结合位点进行预测分析。
1.2.2 vbp2基因阅读框被红色荧光蛋白编码基因rfp准确替代突变体菌株的构建 用红色荧光蛋白编码基因rfp准确替代vbp2基因的突变体的构建采用同源重组的方法。该方法的原理和详细步骤参阅文献[17]。以 A. tumefaciens C58基因组为模板,Q1-F、Q1-R为引物,通过PCR技术扩增vbp2基因翻译起始位点上游526 bp的启动子序列。以带有红色荧光蛋白基因的质粒pCMV6-AN-RFP为模板,用引物Rfp-F、Rfp-R扩增红色荧光蛋白基因rfp。以前两次PCR产物为模板,以Q1-F、Rfp-R为引物,利用重叠延伸PCR技术构建vbp2上游526 bp片段+ rfp片段融合基因。以A. tumefaciens C58基因组为模板,用引物H-F和H-R扩增出vbp2下游1 kb片段。最后利用引物Q1-F和H-R将vbp2上游526 bp片段+rfp片段与vbp2下游1 kb片段通过重叠PCR连接起来。用TaKaRa琼脂糖凝胶DNA回收试剂盒,回收扩增的目的片段和用BamH I和Hind III切过的载体pEX18Km。用T4连接酶将两者4℃过夜连接后转化至大肠杆菌DH5α,经菌落PCR和质粒双酶切鉴定正确后送测序,确定测序结果正确后,将所构质粒pEX18Km-rfp电转至C58感受态细胞,涂布于含卡那霉素抗性的MG/L固体培养基上进行第一次抗性筛选。将长出的单克隆在新的卡那抗性的MG/L固体培养基上纯化,以保证其具有卡那抗性。将纯化的具有卡那抗性的单克隆划线于含5%蔗糖的MG/L固体培养基上,若不长则说明赋予了蔗糖敏感性;选取同时具有卡那霉素抗性和蔗糖敏感性的转化子,划线于含5%蔗糖的MG/L固体培养基上,进行第二次筛选,此时会出现包括野生型和替代突变株,两种类型的单菌落,将长出来的菌落在5%蔗糖的MG/L上纯化一次,再划线于含卡那霉素的MG/L固体培养基上,确认卡那抗性和蔗糖敏感性均已丢失。挑取无卡那霉素抗性和蔗糖不敏感的单菌落进行PCR 鉴定,鉴定成功后将产物送测序。
1.2.3 诱导物对vbp2启动子的诱导及启动子活性的测定 在取得vbp2基因阅读框被红色荧光蛋白编码基因rfp准确替代的突变体菌株后,vbp2启动子的转录活性就可以通过测定其控制的红色荧光蛋白RFP的表达量来表征。具体实验设计是用不同浓度的不同诱导物(乙酰丁香酮AS和鼠李糖Rha)诱导上述突变体菌株。如果vbp2的启动子能被诱导,而表现出不同的转录活性,就可以表达出不同量的红色荧光蛋白RFP,RFP在细胞粗提液总蛋白中的占比就不同。因此,可以用细胞粗提液中单位总蛋白的荧光强度来表示荧光蛋白的表达量。详细实验步骤如下:将上述突变体菌株的单菌落在MG/L培养基中培养至OD600= 0.5-0.7,离心收集菌体后转移至AB蔗糖培养基过渡培养4-5 h,然后分成若干等份,不同的等份分别用不同浓度的不同诱导物进行诱导培养12-16 h。AS的诱导浓度设为0、50、100、150、200和 250 μmol/L。Rha的诱导浓度设为 0、50、100、150、200 和 250 μmol/L。收集经诱导的细胞,用50 mmol/L PBS缓冲液(pH 7.4)洗涤两遍,重悬为总体积10 mL的菌体悬浮液,菌体悬浮液通过调整OD600一致,以保证每份经诱导的菌体悬浮液具有相同的细胞量。用超声波(220 W,3 s on/5 s off,20 min)破碎细胞后,离心获得菌体细胞粗提液。粗提液中的总蛋白浓度采用考马斯亮蓝染料结合法测定[18]。在测得每份待测样品的总蛋白浓度之后,用PBS缓冲液将每份待测样品的总蛋白浓度调成一致。采用Varian Eclipse荧光分光光度计测定每份细菌粗提液的荧光强度,计算单位总蛋白的红色荧光强度。用553 nm波长的光激发红色荧光蛋白的荧光,测定其发射出的574 nm波长的光强[19-21]。
1.2.4 vbp2启动子区被截短的荧光表达载体的构建及被截短的启动子活性的测定 为了确定个别调控位点在启动子区的位置,我们对vbp2启动子区进行截短,得到不同长度的vbp2基因上游片段。将这些不同长度的vbp2基因上游片段用作启动egfp基因表达绿色荧光蛋白GFP的启动子,连接到egfp基因起始密码子的上游。然后将这些连上了不同长度的vbp2启动子区DNA片段的egfp基因分别插入到没有启动子的质粒中,以构建用截成不同长度的vbp2启动子启动egfp基因表达的系列表达质粒。无启动子的质粒构建方法是:以P-F和P-R(均具有Hind III酶切位点)为引物,以pUCA19质粒为模板进行全质粒PCR。之后通过Hind III酶切,T4连接酶连接,得到不含有lacZ’启动子的pUCA19载体,即pUCA19-N质粒。
根据生物信息学分析的结果,在vbp2编码区上游设计多条引物,分别为Q1-F、Q2-F、Q3-F、Q4-F、Q5-F、Q6-F、Q7-F、Q8-F(均具有 BamH I酶切位点),它们都可与引物Q1-R组成一对引物,通过PCR扩增相应的启动子片段,片段大小分别为526 bp、463 bp、260 bp、165 bp、126 bp、97 bp、60 bp、20 bp。然后以eGFP-T质粒为模板,Egfp-F、Egfp-R为引物,通过PCR扩增绿色荧光蛋白egfp基因。利用重叠PCR技术,将上述不同长度的vbp2启动子片段分别与egfp基因相连,构建受不同长度vbp2启动子控制的egfp基因系列表达片段。然后将这些表达片段通过BamH I和Hind III酶位点插入到无启动子质粒pUCA19-N中,即得到了一系列受不同长度vbp2启动子控制的egfp基因表达质粒。在这些egfp基因表达质粒测序正确后,电转至根癌农杆菌GMI9017Δvbp2菌株。让该菌株在诱导物的诱导下由不同长度vbp2启动子控制egfp基因的表达,收集诱导后的细胞,提取细胞粗提液,测定粗提液中单位总蛋白的绿色荧光强度。用488 nm波长的光激发绿色荧光蛋白的荧光,测定其发射出的508 nm波长的光强[22-23]。
2 结果
2.1 vbp2启动子结构的生物信息学预测结果
用公共数据库对vbp2基因启动子区进行生物信息学预测,结果提示,vbp2的启动子区可能存在多个转录调控元件。预测的调控元件及其位点如图1所示。主要有4种,分别是σ38因子结合位点、环腺苷酸受体蛋白结合位点(cap/crp)、fadR和rhaS。如果这些生物信息学的预测结果有一定可靠性的话,vbp2(atu4860)基因的表达调控机制将相当复杂,不仅受多种因素调控,也可能受某种未知的调控机制所调节。鉴于vbp2基因可能存在复杂的调控机制,现阶段我们仅对几种最有可能存在的诱导调控机制进行研究。

图1 生物信息学预测的vbp2基因启动子区可能存在的转录调控元件Fig.1 Feasible transcriptional regulator in the promoter region of vbp2 gene predicted by bioinformatics
2.2 vbp2基因被rfp基因原位替代突变体菌株的构建
为了能够准确地表征vbp2启动子启动转录的活性,我们将vbp2启动子控制的vbp2基因替换成编码红色荧光蛋白的rfp基因,即可通过测定红色荧光蛋白的表达量来表征该启动子的活性。因此,构建vbp2基因被rfp基因原位替代的突变体菌株就很重要。首先需要构建用于根癌农杆菌进行同源重组突变的质粒。图2所示是已经构建好的用于根癌农杆菌进行同源重组突变的质粒pEX18Km-rfp的酶切和PCR鉴定结果。测序结果进一步验证该质粒构建正确。将该质粒电转至根癌农杆菌C58感受态细胞中,通过同源重组整合至根癌农杆菌基因组中。经第一次卡那霉素抗性正向筛选和第二次蔗糖敏感反向筛选,得到无卡那霉素抗性和蔗糖不敏感的原位替代菌株。对所获得的突变体菌株,通过PCR和测序,以确定所获突变体菌株准确无误,并将所获的vbp2基因被rfp基因原位替代的突变体菌株取名为C58vbp2→rfp菌株。

图2 用于进行同源重组构建根癌农杆菌突变体的质粒pEX18Km-rfp的鉴定Fig.2 Identification of plasmid pEX18Km-rfp used for the construction of A. tumefaciens mutant via homologous recombination
2.3 根癌农杆菌粗提液中荧光蛋白浓度与荧光强度的相关性验证
为了明确根癌农杆菌粗提液中的其它成分是否会干扰荧光蛋白的测定,我们将野生型C58(不含荧光蛋白)和替代菌C58vbp2→rfp(含荧光蛋白)的粗提液中的蛋白浓度调成一致,并按不同比例混合,获得含RFP蛋白粗提液占比分别为0%、20%、40%、60%、80%和100%的混合粗提液,并用荧光分光光度计测定这些混合粗提液的荧光强度。粗提液中RFP的比例与荧光强度之间的关系见图3,由图可知,粗提液的荧光强度与粗提液中RFP蛋白的含量呈线性关系,说明粗提液中的其它物质不会干扰RFP蛋白的荧光强度的测定。可以用粗提液的荧光强度来表示荧光蛋白的表达量。

图3 根癌农杆菌粗提液中RFP的含量与粗提液的荧光强度的关系Fig.3 Relationship between fluorescent intensity and RFP content in A. tumefaciens crude extract
2.4 乙酰丁香酮(AS)和鼠李糖(Rha)能够诱导 vbp2启动子启动荧光蛋白的表达
乙酰丁香酮(AS)是最常用来诱导根癌农杆菌转基因的诱导物,同时VBP蛋白的一项重要功能就是将T-复合物招募到四型分泌系统处。因此,我们首先想到的就是AS是否能够调控vbp2启动子的活性。用不同浓度的AS诱导替代菌C58vbp2→rfp,提取这些经不同AS浓度诱导的细胞粗提液,测定这些细胞粗提液的荧光强度。从图4-A可以看出,荧光强度所代表的由vbp2启动子启动表达的RFP的量是受AS诱导调控的。当AS浓度为150 μmol/L时,诱导vbp2启动子启动rfp基因表达的能力最强。
多种单糖也是诱导根癌农杆菌转基因致瘤的诱导物。前面的生物信息学预测,vbp2启动子可能存在一个RhaS蛋白结合位点,在大肠杆菌中RhaS与L-鼠李糖结合后可以激活相关基因的启动子,启动相关基因的表达。因此,我们测试了鼠李糖对vbp2启动子控制的RFP表达的影响。图4-B的结果显示鼠李糖确实对vbp2基因启动子控制的RFP的表达有调控作用。诱导vbp2基因启动子控制的RFP表达的最适鼠李糖浓度为100 μmol/L。

图4 不同浓度的乙酰丁香酮(A)和L-鼠李糖(B)诱导vbp2启动子表达RFP的情况Fig. 4 vbp2 promotor-promoting RFP expressions induced by different concentrations of AS(A)or Rha(B)
2.5 vbp2启动子中AS和Rha调控元件位置的确定
为了进一步确定对AS和Rha诱导做出响应的调控元件在vbp2启动子区的具体位置。我们对vbp2的启动子区域进行了截短,被截短片段的长度和被截去的可能调控元件如图5-A所示。让这些不同长度的vbp2启动子片段控制egfp基因的表达。再将这些带有不同长度的vbp2启动子片段的egfp基因分别插入到没有启动子的质粒pUCA19-N中,获得egfp基因分别受不同长度的vbp2启动子片段控制的系列egfp表达质粒。然后将这些egfp表达质粒分别电转至根癌农杆菌GMI9017Δvbp2感受态细胞,将长出来的菌落进行PCR鉴定,确定这些egfp表达质粒已经分别转入根癌农杆菌GMI9017Δvbp2菌株的细胞内。鉴定结果见图5-B,电泳条带a-h对应的片段大小分别 为 1 246 bp、1 183 bp、980 bp、885 bp、846 bp、817 bp、780 bp和740 bp,与预计相符,说明携带受不同长度的vbp2启动子片段控制的egfp表达质粒的系列根癌农杆菌菌株构建成功。

图5 受不同长度的vbp2启动子片段控制的egfp表达质粒的构建和鉴定。Fig.5 Construction and identification of the egfp expression plasmids controlled by different lengths of vbp2 promotor
在取得了这些携带受不同长度的vbp2启动子片段控制的egfp表达质粒的根癌农杆菌菌株后,我们分别用最适浓度的AS(150 μmol/L)和Rha(100 μmol/L)诱导这些菌株,测定这两种物质诱导不同长度的vbp2启动子启动egfp基因表达绿色荧光蛋白GFP的情况。从图6-A可以看出,当vbp2启动子区缩短至起始密码子上游260 bp处时,受其控制的egfp基因仍然能够被AS诱导表达出绿色荧光蛋白,当vbp2启动子区缩短至起始密码子上游165 bp处时,AS就不能诱导启动子启动egfp基因表达了。由此可以得出响应AS诱导的调控元件位于vbp2上游的165 bp-260 bp区域。图6-B是L-鼠李糖(Rha)诱导这些菌株表达GFP的情况,结果显示,当vbp2启动子区缩短至起始密码子上游165 bp处时,Rha仍然能够诱导GFP的表达,当vbp2启动子区缩短至起始密码子上游126 bp处时,Rha就诱导不了GFP的表达,说明,响应Rha诱导的调控元件位于vbp2上游的126 bp-165 bp区域。
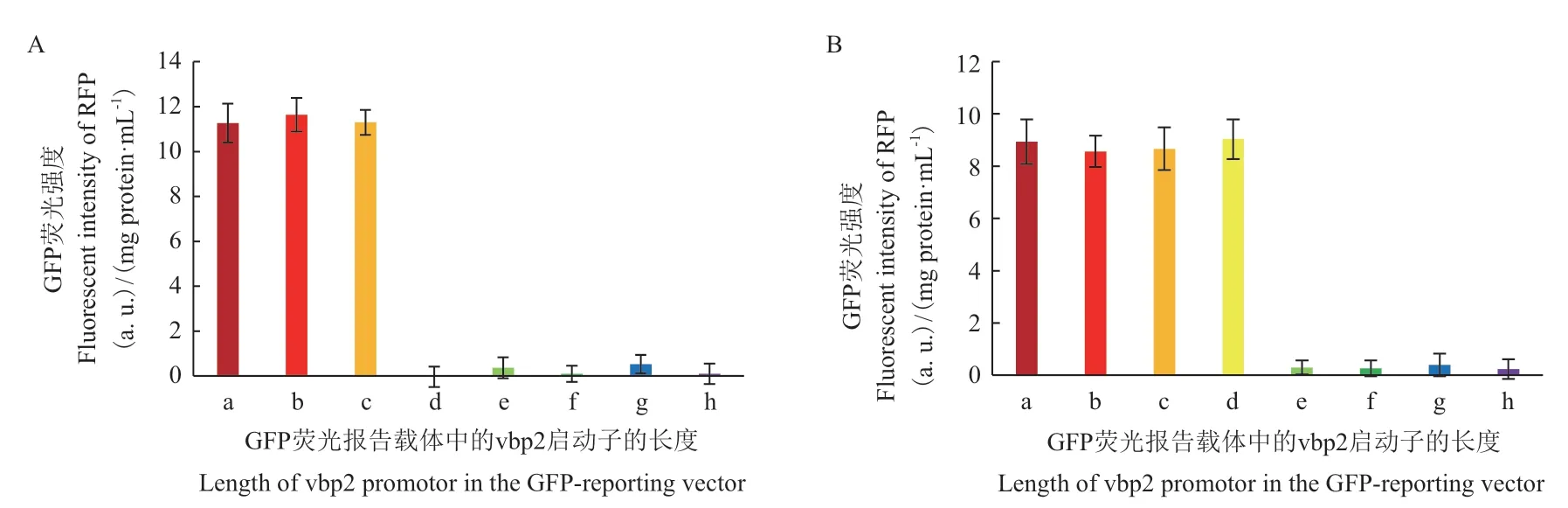
图6 分别以150 μmol/L AS(A)和100 μmol/L Rha(B)诱导不同长度vbp2启动子表达GFP的情况Fig. 6 GFP expression promoted by different lengths of vbp2 promotor under the induction of 150 μmol/L AS(A)or 100 μmol/L Rha(B)
3 讨论
本研究通过构建将vbp2基因的开放阅读框(ORF)准确地替换成rfp基因ORF的菌株来定量地研究vbp2启动子的活性,使红色荧光基因rfp在vbp2基因的原位上由vbp2基因的启动子调控rfp基因的表达,这样不仅可以准确地定量vbp2启动子的活性,而且可以真实地还原根癌农杆菌在接收到诱导物信号之后是如何启动vbp2基因表达的。通过细菌粗提液中荧光蛋白的含量与荧光强度的相关性测定,证明了根癌农杆菌细胞粗提液中的其它物质对荧光强度的测定没有干扰。用AS和Rha两种物质分别对原位替代菌株诱导,发现这两种物质都能诱导vbp2启动子启动rfp基因表达RFP蛋白。
利用截断vbp2启动子区的方法,进一步确定了响应AS诱导的调控元件位于vbp2上游的165 bp-260 bp区域。生物信息学分析预测,vbp2启动子的165 bp-260 bp区域含有一个σ38因子结合位点。因此,我们推测AS可能是通过影响σ38因子与该位点的结合来调控vbp2基因的启动子。在其它革兰氏阴性菌中的研究结果表明,σ38因子主要识别响应环境胁迫的基因的启动子,特别是识别一些与致病性(virulence)有关基因的启动子[24-26]。这些实验结果与AS是诱导根癌农杆菌致病的最好诱导物的推测是非常吻合的[8,27]。AS诱导根癌农杆菌的主要致病基因表达的机制已经很清楚,那就是AS通过VirA/VirG双组分系统诱导位于Ti质粒上的vir基因表达[28-29]。VBP蛋白也影响根癌农杆菌的致病,但vbp2基因位于根癌农杆菌的线型染色体上,不在Ti质粒上[13]。在这里我们证明AS也能诱导vbp2基因的表达,而且可能是通过影响σ38因子与启动子的结合来调控vbp2基因的表达,这与已知的AS诱导调控根癌农杆菌致病基因表达的机理是完全不同的,因此,值得我们进一步研究和探讨。
通过截断vbp2启动子区的方法,我们还确定了响应Rha诱导的调控元件位于vbp2上游的126 bp-165 bp区域。生物信息预测结果显示,该区域可能含有一个rhas位点。在大肠杆菌中,启动子区rhaS位点的调控机制是RhaS蛋白需要与L-鼠李糖结合后才具有活性[30-31]。因此,下一步我们希望能够表达根癌农杆菌的RhaS蛋白,并验证根癌农杆菌的RhaS蛋白是否能与L-鼠李糖形成复合物,并能够特异性地结合vbp2启动子区的这段DNA。此外,现有的研究还表明,RhaS蛋白调控的基因与细菌的碳源利用有关,调节处于不同生存条件(如病原菌的独立生存或作为病原菌生存)下的碳源利用途径[31-32]。我们以前的研究结果推测,VBP蛋白除了招募T-复合物到四型分泌系统处外可能还有其它生物学功能[14,16]。因此,根据这些研究结果和合理分析,我们初步推测,L-鼠李糖诱导vbp2基因表达的另一项功能可能是使根癌农杆菌适应宿主体内的碳源利用途径。当然,还需要更多的实验证据来支持这样的推测。
4 结论
证明了vbp2基因分别受乙酰丁香酮和L-鼠李糖诱导,并确定了两种诱导物的最适诱导浓度。同时,明确了vbp2启动子区受乙酰丁香酮和L-鼠李糖诱导的调控元件分别位于vbp2起始密码子上游165-260 bp和126-165 bp区域。
