一个2A型肢带型肌营养不良家系CAPN3基因的突变分析
2020-05-12徐晓薇张新杰王学韬邹倩倩舒剑波蔡春泉
郑 洁, 徐晓薇, 张新杰, 王学韬, 邹倩倩, 舒剑波, 蔡春泉
肢带型肌营养不良(Limb-girdle muscular dystrophy,LGMD) 是一组以进行性骨盆带和肩胛带肌无力为临床特点的常染色体遗传性疾病[1]。2018年欧洲神经肌肉疾病中心提出新的命名方式[2],将LGMD分为29种亚型,LGMD D1到LGMD D5为常染色体显性遗传;LGMD R1到LGMD R24为常染色体隐性遗传,这些亚型大多与严重先天性肌营养不良相关的基因缺陷有关[3~5]。2A型肢带型肌营养不良(Limb-girdle muscular dystrophy,LGMD2A)又称LGMD R1 calpain3-related,是最常见的亚型之一,占LGMD总数的30%,发病率约1/10万,平均发病年龄17.9岁,由CAPN3基因缺陷导致[6,7]。主要临床表现为近端肌肉无力、肌萎缩,伴翼状肩、肌酸激酶升高等。该病临床表现差异大,临床表现与X染色体连锁的杜氏/贝氏进行性肌营养不良相似,易误诊。目前,肌肉钙蛋白酶-3的缺乏和进一步的CAPN3基因检测为确诊该病的主要方法。在本研究中,使用全外显子测序方法对1个2A型肢带型肌营养不良家系进行基因分析以明确病因,为该家系的遗传咨询提供依据。
1 对象与方法
1.1 对象 先证者(Ⅱ11),男,现58岁。出生时为足月顺产,9岁出现下蹲费力,跑跳困难。19岁出现走路易跌倒,上楼梯需要扶椅,鸭步步态,双肩抬举费力。32岁时仍能够行走,在他人搀扶下可上楼;同年在外院行血液生化检查:血清肌酸激酶560 U/L(正常参考值50~170 U/L),乳酸脱氢酶98 U/L(正常参考值120~250 U/L),谷草转氨酶正常;体格检查:闭眼无力,三角肌、肱二头肌和肱三头肌肌力均为3级;髂腰肌、股四头肌、腓肠肌和胫骨前肌肌力均为2级,腓肠肌无肥大,其余未见异常。肌电图提示肌源性受损;诊断为肢带型肌营养不良。46岁时跌倒致一侧胫腓骨骨折,后使用轮椅。父母非近亲结婚,有家族遗传史:先证者的二姐(Ⅱ5)和三姐(Ⅱ7)有类似病史,先证者二姐(Ⅱ5)现使用轮椅,可在他人搀扶下行走;先证者三姐(Ⅱ7)现使用轮椅,无法行走。先证者兄长和姐姐均已婚,子女均无发病。先证者父亲(Ⅰ1)和长姐(Ⅱ1)已死亡(见图1)。先证者女儿(Ⅲ7)有再次生育愿望,遂来我院行遗传咨询,进行家系基因筛查。本研究获得家系成员的知情同意,并经医院伦理委员会批准。
1.2 方 法
1.2.1 样本 采集收集先证者及其家族成员外周静脉血2 ml于EDTA抗凝管中,4 ℃保存备用。使用康为世纪生物公司的血液基因组提取试剂盒提取基因组DNA,DNA体积100 μl,浓度达10 ng/μl以上,-20 ℃保存备用。
1.2.2 全外显子测序 抽取先证者外周血样1 ml,委托深圳华大基因股份有限公司进行全外显子组高通量测序。测序原始数据使用BWA软件与hg19人类参考基因组进行序列比对,采用GATK 软件进行插入缺失、单核苷酸多态性位点等分析,用Annovar软件和千人基因组数据库、dbSNP、OMIM等数据库进行注释,利用Polyphen2和SIFT等软件进行蛋白功能预测。
1.2.3 Sanger测序验证 根据全外显子测序结果,采用Sanger测序对先证者及其家系成员进行验证。PCR扩增家系中其余2例患者和正常家系成员基因。针对CAPN3基因c.1194-9A>G(NM_000070.2)突变设计引物,上游引物为5’-GCCACCCTCTTTTCATCCTCC-3’,下游引物为5’-TGTTCCCACAGTTTCCTGCTTC-3’,Tm值为58 ℃。针对CAPN3基因c.1437C>T(NM_000070.2)突变设计引物,上游引物为5’-TGTAGGGAATAGAAATAAATGG-3’,下游引物为5’-CCAGGAGCTCTGTGGGTCA-3’,Tm值为60 ℃。PCR反应条件为:95 ℃预变性10 min,95 ℃ 30 s,58 ℃/60 ℃ 30 s,72 ℃ 40 s,共35个循环,最后72 ℃延伸10 min。PCR产物使用1.5%琼脂糖凝胶电泳检测。使用康为世纪生物公司的琼脂糖凝胶DNA回收试剂盒纯化DNA后,送至北京金唯智生物科技公司进行Sanger测序。测序结果用Chromas软件与GenBank中参考序列比对,确定突变位点。应用在线软件Human Splicing Finder(http://www.ummd.be/HSF/)进行基因剪接位点预测。
2 结 果
2.1 全外显子测序结果 结果显示先证者CAPN3(NM_000070.2)基因存在复合杂合突变。第9内含子存在c.1194-9A>G杂合突变,生成一个剪切位点突变。在第5外显子上存在另一个杂合突变,c.1437C>T(p.ser479=),第479位的遗传密码子由AGC变成AGT,氨基酸丝氨酸未发生改变的同义突变。检索ClinVar数据库,c.1194-9A>G变异为已经报道过的致病性变异;c.1437C>T(p.ser479=)变异未见相关文献报道,该变异为新突变。
2.2 Sanger测序验证结果 Sanger测序结果与全外显子测序结果一致。结果显示先证者(Ⅱ11)CAPN3基因c.1194-9A>G变异来源于母亲(Ⅰ2),c.1437C>T(p.ser479=)未在母亲测序结果中发现。家族中患者(Ⅱ5、Ⅱ7)也存在相同的复合杂合突变,其四姐(Ⅱ9)和女儿(Ⅲ7)为c.1194-9A>G变异携带者,先证者的女婿(Ⅲ8)未检测到上述位点变异(见图2)。
2.3 生物信息学分析 应用在线软件Human Splicing Finder(http://www.ummd.be/HSF/)评估该变异可能影响剪切,产生新的剪接位点(见图3)。

A:先证者母亲(Ⅰ2)存在c.1194-9A>G杂合变异;B:先证者二姐(Ⅱ5)存在c.1194-9A>G和c.1437C>T的复合杂合突变;C:先证者三姐(Ⅱ7)存在c.1194-9A>G和c.1437C>T的复合杂合突变;D:先证者四姐(Ⅱ9)存在c.1194-9A>G杂合变异;E:先证者(Ⅱ11)存在c.1194-9A>G和c.1437C>T的复合杂合突变;F:先证者女儿(Ⅲ7)存在c.1194-9A>G杂合变异;G:先证者女婿(Ⅲ8)无该位点变异
图1 先证者家系图
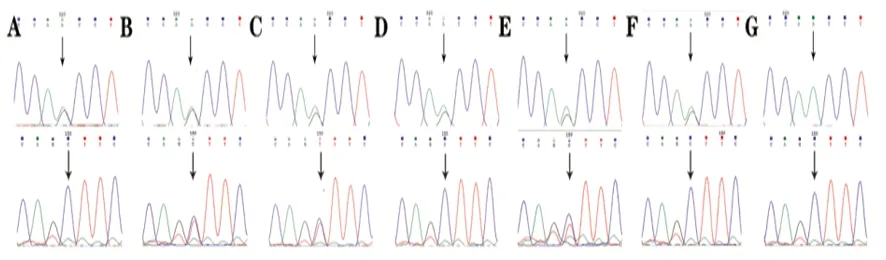
图2 CAPN3基因两个突变位点的Sanger测序结果

图3 生物信息学软件预测结果图
3 讨 论
LGMDs是一组累及近端肌肉,主要表现为骨盆带肌和肩胛带肌无力的常染色体遗传性疾病。发病率约1/10万,从2岁至55岁均可发病,平均发病年龄17.9岁。患者自发病至依赖轮椅的平均病程为15.2 y,平均年龄为35.2岁[6]。该病具有高度异质性,肌无力的进展通常是对称性的,但病情严重程度各不相同[8]。LGMD2A的主要临床特点是进行性肌无力,可表现为跑跳困难、翼状肩、异常步态等,在病程晚期可有呼吸肌受累导致呼吸衰竭[9,10]。该病的诊断主要依靠临床表现、肌肉活组织病理学检查以及基因检测确定突变,这同时也是进行遗传咨询的关键[11]。
LGMD遗传模式分为常染色体隐性遗传和常染色体显性遗传,最常见的LGMD2A型是由CAPN3基因突变引起的常染色体隐性遗传病[12],目前已经发现了约500个变异,也有报道CAPN3基因内21个碱基对缺失可导致常染色体显性遗传[5]。CAPN3基因位于染色体15q15区,该基因由24个外显子组成,编码的产物为calpain3[13]。Calpain由821个氨基酸组成,是一种肌肉特异性非溶酶体半胱氨酸蛋白酶,参与肌肉再生、肌膜重构、细胞骨架调控和钙稳态,与多种关键骨骼肌蛋白的分解和裂解相关,尤其是与肌原纤维蛋白骨架构成相关的蛋白,例如肌联蛋白、细丝蛋白C、黏着斑蛋白等[14~16]。CAPN3基因突变导致这些蛋白活性的丧失,与LGMD2A的发病机制有关。
本研究中,先证者9岁起病,以骨盆带肌受累为主,出现下蹲费力、跑跳困难,无翼状肩和腓肠肌肥大,血清肌酸激酶升高,肌电图提示肌源性受损。先证者于20余年前被诊断为贝氏进行性肌营养不良,但是根据家系调查研究,本家系在Ⅱ代中出现了2名女性患者,后一代男性均无患者,不符合X连锁隐性遗传的遗传模式。全外显子测序结果显示先证者CAPN3基因存在一个已知的致病变异c.1194-9A>G,同时检测到另一同义突变c.1437C>T(p.ser479=),该同义突变在千人基因组及gnomAD数据库中,东亚人群的频率为3‰和2.5‰,全人群的频率为6‰和1‰,不属于SNP位点。故结合其临床表现和病史,可确诊为LGDM2A。利用Sanger测序对家族成员进行验证,发现家族中患者(Ⅱ5、Ⅱ7)存在相同的复合杂合突变, c.1194-9A>G变异来源于先证者母亲,其四姐(Ⅱ9)和女儿(Ⅲ7)为c.1194-9A>G变异携带者。1194-9A>G突变导致新的剪接位点位于正常位点的下游9 bp处,使得第9内含子最后8个碱基得以保留,产生一个新的增强子,这与第7内含子中Alu元件的多次插入有关,使得转录本中前7个外显子缺失[17]。c.1437C>T(p.ser479=)变异在家族中患者(Ⅱ5、Ⅱ7、Ⅱ11)均携带,根据美国ACMG遗传变异分类标准与指南[18],该变异符合致病性证据PM2:ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的变异;PM3:在隐性遗传病中,在反式位置上检测到致病变异,并已通过患者父母验证;PP1:突变与疾病在家系中共分离;PP3:预测该变异可能影响剪切;PP4:变异携带者的表型或家族史高度符合某种单基因遗传疾病。符合“2个中等(PM2、PM3)和≥2个支持(PP1、PP3、PP4)”,根据以上证据认为该变异为可能致病的。
综上所述,本研究中我们对一个LGMD2A型的家系进行基因突变的分析,发现先证者存在CAPN3基因c.1194-9A>G和c.1437C>T(p.ser479=)的复合杂合突变,通过家系研究及生物信息学预测,初步证实了c.1437C>T(p.ser479=)为一导致剪接位点突变的新突变,丰富了CAPN3基因的突变谱,为基因诊断和遗传咨询提供了依据。
