Wilson病疑似自身免疫性肝炎1例及文献复习
2019-05-08
肝豆状核变性由Wilson在1912年首先报道,故又称为Wilson病,是由ATP7B基因突变引起铜在肝脏及其他组织中沉积,导致肝脏、神经和精神系统异常的一种常染色体隐性遗传的罕见疾病,经Wilson病诊断积分≥4分可确诊。临床上,成人型Wilson病常难以与自身免疫性肝炎(AIH)鉴别,导致患者无法得到准确而及时的治疗。现报道上海交通大学医学院附属仁济医院诊治的1例Wilson病患者,并对此罕见疾病进行文献复习。
1 病例资料
患者女性,30岁,因“发现肝功能反复异常1年半”于2017年9月18日入院。患者于2016年3月17日孕16周时发现肝功能异常:丙氨酸氨基转移酶(ALT)113 U/L,天冬氨酸氨基转移酶(AST)154 U/L,谷氨酸转肽酶(GGT)48 U/L,碱性磷酸酶(ALP)107 U/L;乙型肝炎两对半:HBsAg阳性,HBeAb阳性,HBcAb阳性;HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎抗体均为阴性。EB病毒(EBV)、巨细胞病毒(CMV)抗体均为阴性。腹部B超未见明显异常。外院诊断为“妊娠合并肝损、非活动性HBsAg携带者”,予复方甘草酸苷、腺苷蛋氨酸保肝治疗,肝功能改善后出院(ALT 47 U/L,AST 72 U/L)。2016年9月14日孕38周顺产一名男婴。产后出现发热,于外院检查发现氨基转移酶进行性升高:ALT、AST、GGT、ALP分别升至324 U/L、433 U/L、88 U/L、158 U/L;总胆汁酸(TBA)15.7~27.6 μmol/L;HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎病毒标志物均阴性。EBV衣壳抗原IgA抗体阳性,EBV-DNA 8.910 7 copies/mL。腹部彩超未见明显异常。遂诊断为“EBV感染伴肝损、非活动性HBsAg携带者”,予更昔洛韦抗病毒、复方甘草酸苷、腺苷蛋氨酸保肝治疗,肝功能改善后出院(ALT 89 U/L,AST 109 U/L )。2017年9月10日外院复查腹部B超提示肝硬化,脾大。肝功能再次异常:ALT 125 U/L,AST 144 U/L,GGT 72 U/L,ALP 108 U/L。HBsAg阳性,HBeAb阳性,HBcAb阳性,HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎病毒标志物均阳性。EBV衣壳抗原IgA抗体阳性,EBV-DNA阴性,CMV-Ab阴性,自身免疫性抗核抗体谱阴性,IgG 17.8 g/L,IgM 2.17 g/L。于2017年9月15日行肝穿刺活组织病理检查,结果显示:小叶结构紊乱伴假小叶形成,门管区、纤维间隔内中-重度慢性炎性反应伴中度界面性肝炎(图1A),炎性细胞主要为淋巴细胞,可见浆细胞及中性粒细胞,部分肝细胞呈“玫瑰花环”样排列(图1B)。CK7免疫组化检查示门管区、纤维间隔胆管明显增生。铜染色可疑阳性。病理诊断为:慢性肝炎,肝硬化形成(G3,S4)。门诊诊断为:(1)肝炎后肝硬化,自身免疫性肝炎?(2)非活动性HBsAg携带者。患者既往5年前婚检时发现乙型肝炎小三阳、肝功能正常,腹部B超无异常;有轻度贫血史,未诊治;有桥本甲状腺炎病史;否认长期服药史、否认吸烟饮酒史。2012年顺产一足月男婴;2014年因前置胎盘,孕6月时流产,这两次孕期均无肝功能异常史。2016年孕38周时顺产一男婴,妊娠期有肝功能异常及TBA升高史。患者为独生女,父母否认慢性肝炎史,叔叔因“肝硬化”于30岁时病逝。患者入本院后查体无明显异常,继续完善相关辅助检查。自身免疫性肝病抗体:抗核抗体(ANA)1∶100,抗纤维肌动蛋白抗体(F-actin)35.19 IU,余抗体阴性。免疫球蛋白组合:IgG 17.2 g/L(7~16 g/L), IgM 1.61 g/L(0.4~2.3 g/L)。铜蓝蛋白(CPN)0.09 g/L(0.2~0.6 g/L),血清铜8.9 μg/dL(11.8~39.3μg/dL),尿铜193.4 μg/24 h(0~60 μg/24 h)。颅脑MRI未见异常。基因检测:ATP7B基因复合杂合突变(p.Thr935Met,p. Ser975Phe)。眼科会诊未见Kayser-Fleischer(K-F)环。遂确诊为:Wilson病,非活动性HBsAg携带者。予锌剂口服治疗(每日3次,每次50 mg),3个月后随访肝功能恢复正常。
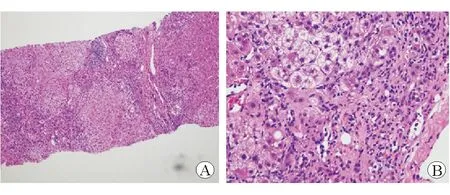
图1肝脏活组织病理检查 苏木精-伊红染色A×100B×200
2 讨论
Wilson病是由细胞铜转运缺陷,导致铜在肝脏及其他组织(包括脑)内蓄积,从而导致肝脏、神经系统和精神系统的异常表现[1]。该病分布于世界各地,人群中的患病率约为3万分之一[2]。男性和女性的患病率大致相同,但女性较男性更易发生急性肝衰竭[3-5],而男性较女性更易发生神经精神性疾病[6]。Wilson病是一种常染色体隐性遗传病,由于13号染色体上编码铜转运蛋白ATP7B的基因发生突变[7],导致铜与CPN的结合以及向胆汁中的排泄减少,从而在肝脏中蓄积。过量的肝铜使自由基产生增多,对细胞器的氧化损伤增加,导致肝细胞损伤。肝脏铜含量增加以及肝细胞损伤导致铜释放入血,因此推测血清游离铜(不与血清CPN结合)增加是肝外铜沉积和随后在脑及其他组织中产生毒性的原因。
大多数Wilson病患者在5岁至35岁之间被确诊。Wilson病的临床表现主要在肝脏、神经系统和精神疾病方面,许多患者具有这些症状的组合。肝脏表现包括伴有Coombs阴性溶血性贫血的急性肝功能衰竭、急性肝炎、慢性肝炎、肝硬化、肝脂肪变性和无症状的肝脏生化异常。神经系统表现广泛,大多数神经性Wilson病的患者具有构音障碍和(或)运动障碍,依据此诊断该病具有一定挑战性。其他临床表现有Coombs阴性溶血性贫血和K-F环。Wilson病的辅助检查中尿铜、CPN、K-F环的敏感度和特异度均不高。该病的肝脏活组织检查也呈各种类型的病理改变,包括脂肪变、气球样变、Mallory小体、界面炎、桥接坏死、肝硬化,只有10%的患者肝铜染色为阳性。
Wilson病的诊断尚存在这些困难:(1)发病年龄悬殊(3岁~80岁);(2)多脏器累及;(3)初发症状多样,临床表现复杂多变(从轻微肝功能异常到暴发性肝衰竭);(4)辅助检查的敏感度和特异度不高;(5)基因检测尚未普及;(6)临床医生对该疾病的认识不足。因此,对于具有疑似Wilson病临床特征的患者,应首先检测肝功能、血常规、血清CPN和血清铜水平、眼裂隙灯检查和24 h尿铜。这些检查的结果决定了进一步行肝穿刺或组织病理检查和基因检测的必要性。目前,Wilson病的诊断依据2001年莱比锡国际会议制定的诊断评分系统[8]。本病例为中年女性,以反复肝功能异常为临床表现,其中血清CPN<0.1 g/L,尿铜>2倍正常值上限(ULN),肝组织铜染色可疑阳性,基因检测发现复合杂合子突变,可评8~9分(见表1),故确诊为Wilson病。
自身免疫性肝炎(AIH)是一种慢性炎性肝病,女性多发,分布年龄广,通常以自身抗体和血清球蛋白水平升高为特征,临床表现变化大(从无症状到急性肝衰竭)。其诊断基于排除其他慢性肝病后的特征性血清学和组织学表现(包括界面性肝炎、汇管区和小叶内淋巴浆细胞浸润和肝细胞玫瑰花结)。AIH初始治疗通常采用糖皮质激素,大部分患者对激素应答良好。在早期的诊治中,因本病例符合AIH的简化诊断标准[9],评分7分可确诊,故极易误诊为AIH;而通过AIH综合诊断积分系统[10],评分14分,只能得出AIH可能的结论。患者后期锌剂单药治疗后肝功能改善也说明了本例患者的病因是一元论而非二元论。
总之,Wilson病十分罕见,患者的肝脏活组织病理检查及血清免疫学特点与典型AIH相似。鉴于本病例依据AIH简化诊断标准无法鉴别Wilson病,因此临床上应高度注意:(1)对3岁~80岁不明原因的肝损伤、肝硬化患者,应常规排查Wilson病;(2)诊断AIH前应除外Wilson病,特别是激素应答不良、合并Coombs阴性溶血性贫血或神经系统症状的患者;(3)推荐CPN、24 h尿铜和K-F环作为Wilson病检查的首选指标,而肝组织病理检查并非诊断的充分必要条件;(4)K-F环阴性、CPN正常均不能除外Wilson病,CPN降低也不能确诊Wilson病;(5)Wilson病患者应终身治疗,定期随访评估。

表1 患者的Wilson病诊断评分
注:≥4分为诊断确立;3分为诊断可能,需更多检查;≤2分为诊断非常不可能;患者总得分为8~9分;a或脑MRI的典型异常或UWDRS评分;b如无定量肝铜可用
