伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病一家系中NOTCH3基因的新突变
2019-04-03杨程程滕军放丁雪冰王雪晶
杨程程,滕军放,丁雪冰,王雪晶
郑州大学第一附属医院神经内科 郑州 450052
伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, CADASIL)是一种单基因遗传性脑小血管病,是由位于人类19号染色体短臂上的NOTCH3基因突变所致[1-3]。CADASIL一般中年起病,与传统的脑血管病不同,其通常无明确的脑血管病高危因素,以反复脑卒中发作为主要表现,同时可伴随认知障碍、痴呆、精神情绪紊乱及有先兆的偏头痛等。继2000年第1例CADASIL家系在中国被报道之后,共有20多种NOTCH3基因突变逐渐被发现[4],全世界成千上万CADASIL家系在不同种族中被发现,然而由于不同种族及地域CADASIL的临床表现、基因突变类型等差异较大,这类疾病仍经常被忽视和误诊[5]。MRI上可以发现较多皮质下缺血灶及广泛的白质病变,是诊断CADASIL脑部血管损害的重要手段。目前,CADASIL的诊断除临床症状和影像学检查之外,最重要的是基因检测及病理学组织活检。组织活检能够检验出血管壁内NOTCH3基因编码蛋白的存在及平滑肌细胞膜和膜外基质之间颗粒状嗜锇物质的存在[6]。由于皮肤组织较中枢神经组织容易取材,且皮肤通常也可以发现颗粒状嗜锇物质的存在,因此皮肤活检是此病病理学诊断中经常采用的检测方法[7-9]。由于我国基因诊断技术应用的局限性,经病理及基因检测确诊的CADASIL家系在国内较少,且目前关于NOTCH3基因突变热点的报道多位于第3、4、11、12、13、14、18号等外显子[10]。本文确诊了1例NOTCH3基因第6号外显子基因突变所致的CADASIL家系,报道如下。
1 资料与方法
1.1临床资料2018年5月于郑州大学第一附属医院神经内科住院的1例疑似CADASIL患者,家系图谱见图1。患者男,49岁,慢性起病,以“反应迟钝1 a余”为主诉入院;1 a余前逐渐出现反应迟钝、记忆力下降、表情减少、主动与人沟通能力下降。5个月前上述症状加重,且出现理解力下降、右侧肢体无力、步态不稳、言语不利,至北京某医院行脑脊液免疫学相关检查未见异常,栓子检测、发泡试验均未见异常。
查体:意识清楚,精神一般,语言欠流利,记忆力减退,理解力差,计算力差,判断力正常,右利手。双侧额纹对称,双侧眼裂对称,闭目有力,右侧鼻唇沟变浅,示齿口角左偏,鼓腮不漏气。伸舌充分,舌尖位置右偏,无舌肌萎缩及肌束震颤。无肌肉萎缩及假性肌肥大。四肢肌张力正常,左侧肢体肌力5级,右侧肢体肌力5(-)级。右上肢腱反射稍活跃,左上肢腱反射正常。双下肢腱反射减弱。双侧指鼻试验稳准,右侧跟膝胫试验欠稳准,Romberg征阴性。无不自主运动,步态正常。右侧巴氏征阴性,左侧巴氏征可疑阳性。双侧唇反射阳性,双侧掌颏反射阳性。
影像学检查:头颅MRI示左侧侧脑室后角急性梗死灶。双侧额顶叶、双侧半卵圆中心、放射冠、双侧丘脑及基底节区、桥脑缺血灶及腔梗。头颅MRA示颅内动脉粥样硬化,右侧椎动脉颅内段纤细。头颈联合CTA示主动脉弓局部管壁增厚并点状钙斑影,管腔未见明显狭窄;右侧椎动脉较对侧纤细,硬膜内段为著。结果见图2。
既往史:吸烟史10余年,无高血压、糖尿病、高血脂病史。
该家系中先证者父亲生前有该病临床症状,先证者及其兄有明确的临床症状,其他现存亲属尚无临床症状。先证者兄(Ⅲ5),52岁,以“反应迟钝5 a”为主要临床症状,此外疑似伴随认知功能障碍,拒绝就诊。

■男性患者;□正常男性;○正常女性;/已死亡;先证者
图1先证者家系图谱
1.2皮肤活检告知先证者并获得其知情同意后,取其胫骨前部一块约1 cm×1 cm大小的皮肤组织进行活检。对标本进行固定、乙醇脱水、浸蜡包埋、切片、贴片、HE染色,光镜与电镜下观察。
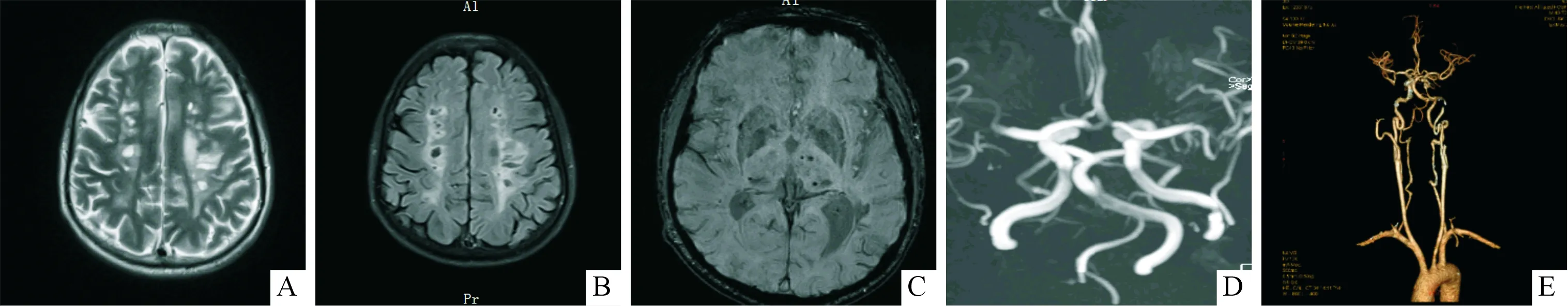
A:MRI T2加权像示双侧顶叶、双侧侧脑室旁多发斑片状高信号;B: T2 FLair像上双侧顶叶、双侧侧脑室后角多发软化灶;C:SWI像上双侧丘脑、右侧基底节区、右侧颞顶叶、左侧额叶少量含铁血黄素沉积或微出血灶;D:脑MRA未见明显局限性狭窄或闭塞;E:CTA示主动脉弓局部管壁增厚并点状钙斑影,管腔未见明显狭窄,右侧椎动脉较对侧纤细,硬膜内段为著
图2先证者头颅影像学表现
1.3高通量捕获测序
1.3.1 全基因组DNA的提取与样本处理 在知情同意的情况下,采用EDTA抗凝采血管采集先证者、先证者兄、先证者子女外周静脉血5 mL,-20 ℃保存。然后采用血液提取试剂盒提取血液DNA,取1.5 μg DNA样本进行超声打断,得到150~200 bp的DNA片段。
1.3.2 DNA文库构建及样本捕获 采用KaPa末端修复酶修复DNA,用AMPure磁珠进行纯化,并采用KaPa Atailing试剂在DNA 3’端添加A碱基,然后采用KaPa DNA Ligase将Adapter连接到DNA片段上,最后进行文库模板量平衡的线性扩增,扩增体系为25 μL。反应条件:2×KaPa HiFi HotStart Ready Mix 12.5 μL,PCR Primer Premix 1 μL,Adapter-ligated library DNA 10 μL,加水至总体积25 μL。反应程序如下:98 ℃ 2 min;98 ℃ 30 s,65 ℃ 30 s,72 ℃ 30 s,8个循环;72 ℃ 4 min。
真空浓缩DNA文库样本进行杂交捕获。反应条件为:DNA文库样本10 μL 95 ℃加热5 min后,加热SureSelect杂交缓冲液且置65 ℃,5 min后捕获探针文库混合液,65 ℃孵育2 min。取14 μL的杂交缓冲液加入探针文库混合液中,再将全部样本文库溶液加入,65 ℃孵育 24 h。取50 μL链霉亲和素(SA)包被的Dynal磁珠(Invitrogen公司产品),用300 μL结合缓冲液洗涤3次后200 μL结合缓冲液重悬Dynal磁珠,并将完成捕获反应的样品全部加入,放入翻转混匀仪中室温混匀孵育30 min。置磁力架静置,待磁珠完全吸附至侧壁后去上清。200 μL洗涤缓冲液1重悬,放入翻转混匀仪中室温混匀孵育15 min。去上清后加200 μL 65 ℃预热的洗涤缓冲液2重悬,置恒温混匀仪中孵育10 min,重复3次。用40 μL的洗脱缓冲液重悬磁珠。取20 μL重悬磁珠,使用Agilent公司的试剂盒提供的试剂进行PCR反应。PCR反应条件为:98 ℃预变性2 min;98 ℃变性30 s;65 ℃复性30 s;72 ℃延伸30 s,14个循环;72 ℃再延伸4 min,4 ℃保存。PCR产物采用Ampure磁珠进行纯化,并进行Qubit定量。
1.3.3 测序 取捕获后的DNA样本进行Illumina HiSeq 2500高通量测序。参考美国医学遗传学和基因组学学院相关指南进行数据解读和生物信息学分析,并与人类基因突变数据库(HGMDpro)进行比对。
2 结果
2.1皮肤组织活检结果光镜下,皮肤组织、表皮基底层色素增多,真皮浅层小血管周围少许淋巴细胞浸润(图3A),浅、深层部分小血管壁增厚(图3B)。电镜下,小动脉管壁可见平滑肌细胞(图3C),平滑肌细胞肌膜下密斑丰富(图3D),平滑肌细胞膜和膜外基质间可见多个嗜锇颗粒沉积,电子密度不一(图3E)。

A、B:光镜(HE,×200);C、D、E:电镜
2.2基因检测结果见图4。NOTCH3基因第6号外显子区域发现一处杂合突变点:c.811T>C,导致氨基酸改变(半胱氨酸>精氨酸)。HGMDpro数据库未见报道。

图4 NOTCH3基因 第6号外显子区的杂合突变(c.811T>C)
3 讨论
CADASIL是一种常染色体显性遗传性脑动脉病。其临床特征及影像学表现被逐步认识 。典型的临床特征包括30岁左右出现的先兆性偏头痛,伴有皮质下梗死并在40岁左右出现相关症状,50岁左右开始出现认知功能下降。CADASIL的头部MRI表现与年龄和疾病阶段相关,表现为T2加权像和T2 Flair像上的对称的、广泛的大面积高信号,DWI像上可出现弥散受限的高信号病灶,即急性或亚急性脑梗死灶;白质病变经常累及前颞叶、外囊、额上回和颞极,这些特殊部位的病变对诊断具有高度的敏感性和特异性,但在亚洲人群中,颞叶病变较为少见[10]。本文家系中2例患者的发病年龄、先证者的影像学表现与之前报道的基本一致。
CADASIL的病理生理机制主要是由于变异的NOTCH3蛋白聚集体(颗粒状嗜锇物质)沉积在中等大小的动脉血管壁的平滑肌细胞表面,造成血管狭窄闭塞,进而导致脑缺血发作。这种沉积通常为全身性、系统性的,但以脑血管壁上的沉积更显著,在脑血管以外的区域,如皮肤和肌肉血管,也可以发现颗粒状嗜锇物质的存在[1-3]。NOTCH3基因突变是CADASIL发病的主要遗传机制。迄今为止,已发现数千个CADASIL家系,其中200多种不同的NOTCH3基因变异已被证实,而在中国大陆的CADASIL家系中,NOTCH3基因突变热点主要位于3、4、11、12、13、14号外显子[11-12]。目前大部分的研究[13]认为在CADASIL家系中,NOTCH3基因突变导致表皮样生长因子重复区域内半胱氨酸残基的重复或丢失,从而导致其编码的跨膜蛋白发生结构和功能的变化。在本文报道的CADASIL家系中,NOTCH3基因第6号外显子的突变(c.811T>C)使半胱氨酸变为精氨酸,破坏了二硫键的原有结构,使其编码的跨膜蛋白失去相应功能,变异的NOTCH3蛋白聚集体(颗粒状嗜锇物质)沉积在中等大小的动脉血管壁的平滑肌细胞表面,造成血管狭窄闭塞,进而导致脑缺血发作。另外皮肤组织活检也进一步证实了这种颗粒状噬锇物质不仅在脑血管内沉积,在皮肤血管内也有沉积。与之前的发现的突变类型不同,本文主要报道了1个中国CADASIL家系中新的NOTCH3基因杂合突变类型(c.811T>C),该突变位点位于第6号外显子,HGMDpro数据库尚未见报道。
综上所述,作者的研究为CADASIL的分子遗传机制提供了新的证据,若进一步在大样本中得到证实,则可能预测这一类基因突变导致的临床表型,提高CADASIL的临床诊治率。
