基于Illumina测序分析青海土族牙周病患者可疑致病微生物的组成差异和种群结构※
2018-12-14李志艳朱德锐
陈 筠,李志艳,赵 翀,刘 静,张 欣,朱德锐*
(1.青海大学附属医院 口腔内科 西宁 810001;2.青海大学医学院 基础医学研究中心 810016)
目前利用高通量测序技术(High throughput sequencing)开展微生物多样性检测、宏基因组学和微生物代谢组学方法来做口腔致病微生物的组成差异、种群结构研究,是国内外研究的热点[1-3]。
研究者通过对海拔2 500 m以上的2 400名世居者及移居者进行牙周病学普查,发现土族移居者牙周病发病率高达98.7%,高于其他民族[4-6]。研究显示,高原地区(海拔4500m)的居民口腔疾病发病率及病程进展速度显著高于平原地区,牙周病的高发可能与其特殊的致病微生物的多样性和组成有关。然而针对高原地区牙周病的致病微生物的研究报道相对较少,且结论不一。因此本研究以青海土族人群为研究对象,采用16S rRNA基因高通量测序技术初步探究青海土族牙周病患者口腔可疑致病微生物的组成差异和种群结构,为后期牙周病的预防、诊断与治疗提供依据。
1 材料与方法
1.1 研究对象选取和唾液采集
选取青海省互助土族自治县威远镇小庄村的土族居民牙周病样本10例。根据第三次全国口腔流行病学调查标准[7]收集慢性牙周炎患者样本,牙周袋深度(PD)≥4 mm或附着丧失量(AL)≥2 mm,所有的选取对象无侵袭性牙周炎、口腔粘膜病、溃烂或智齿冠周炎等干扰本研究结果的疾病。就诊前6个月内未做口腔治疗。采样前先嘱受试者轻漱口,去除口腔中的食物残渣,停止吞咽1 min后让唾液(1.5mL)自行流入5 mL无菌EP管中,标记样本为T1~T10,冰浴运输,2 h内检测完毕。
1.2 基因组DNA的提取及质量控制
样本于0.22 μm细菌滤膜上进行真空抽滤,将滤膜剪碎放入DNA试剂盒(QI Aamp Fast DNA Stool Mini Kit),参照试剂盒步骤提取基因组DNA,用2.0%琼脂糖凝胶电泳分析环境样本DNA完整性。DNA质量纯度检测分析采用微量检测仪Microplate Reader(USA,MD公司),合格的总DNA样品保存于-80 ℃冰箱。
1.3 PCR扩增和测序
使用16S rRNA基因通用引物341F(5′-CCTACGGGNGGCWGCAG-3′)和785R(5′-GACTACHVGGGTATCTAATCC-3′)进行V3-V4目的基因扩增细菌(ABI Gene Amp 9700,USA),PCR反应条件:95 ℃ 5 min,94 ℃ 45 s,55 ℃ 30 s,72 ℃ 90 s,共35个循环,最后在72 ℃下延伸5 min。采用Axyprep DNA Gel Extraction Kit(Axygen scientific,USA)行PCR扩增产物纯化。采用Qubit®2.0荧光计(Invitrogen,USA)行DNA精准定量分析。检测合格的PCR纯化产物行16S rRNA基因高通量测序(MiSeq,美国ABI公司),由上海惠研生物科技有限公司完成,每个样本获得的双端序列数据为2~3万个reads。根据测序reads之间overlap关系进行拼接(merge),同时对reads质量和merge效果进行质控过滤(Q30>80%),根据序列首尾两端的barcode和引物序列区分样品得到有效序列。
1.4 高通量数据统计分析
使用QIIME1.8.0软件对原始数据进行处理,在相似度97%水平上聚类出可操作分类单元OTU(Operational taxonomic unit),进行OTU picking和物种注释分析。使用alpha稀释曲线(Alpha rarefaction.py)计算样品的菌群多样性香农指数(Shannon)和菌种丰富度指数(Chao1)以及谱系多样性(PD whole tree)和观察物种(Observed species)种数,以及基于OTU进行主成分分析(PCA,principal component analysis)。根据分类学分析结果(纲、属与种)分析微生物的群落结构、物种组成比例及丰度。
2 结果
2.1 测序数据统计分析
通过Illumina MiSeq平台进行土族口腔样本高通量测序,获得细菌的总有效序列Sequences为107 861条,平均长度为253 bp。在相似度97%水平上聚类可操作分类单元,并进行OTU picking,获得青海土族牙周病患者口腔微生物的物种注释OTUs数目为156个,不同样本测序的可观察物种和多样性数据结果见表1。土族牙周病的可观察物种OTUs有30~88个、丰度指数Chao1介于38.21~98.66、多样性Shannon指数介于0.29~3.80,表明牙周病患者口腔内具有复杂的微生物生态结构。此外,牙周病组样本间各多样性指数因样本个体的不同而存在差异。随着样本测序深度的增加,覆盖度≥99.70%,表明测序足以可信地反映口腔微生物的存在现状。
表1土族口腔微生物测序统计和多样性分析表
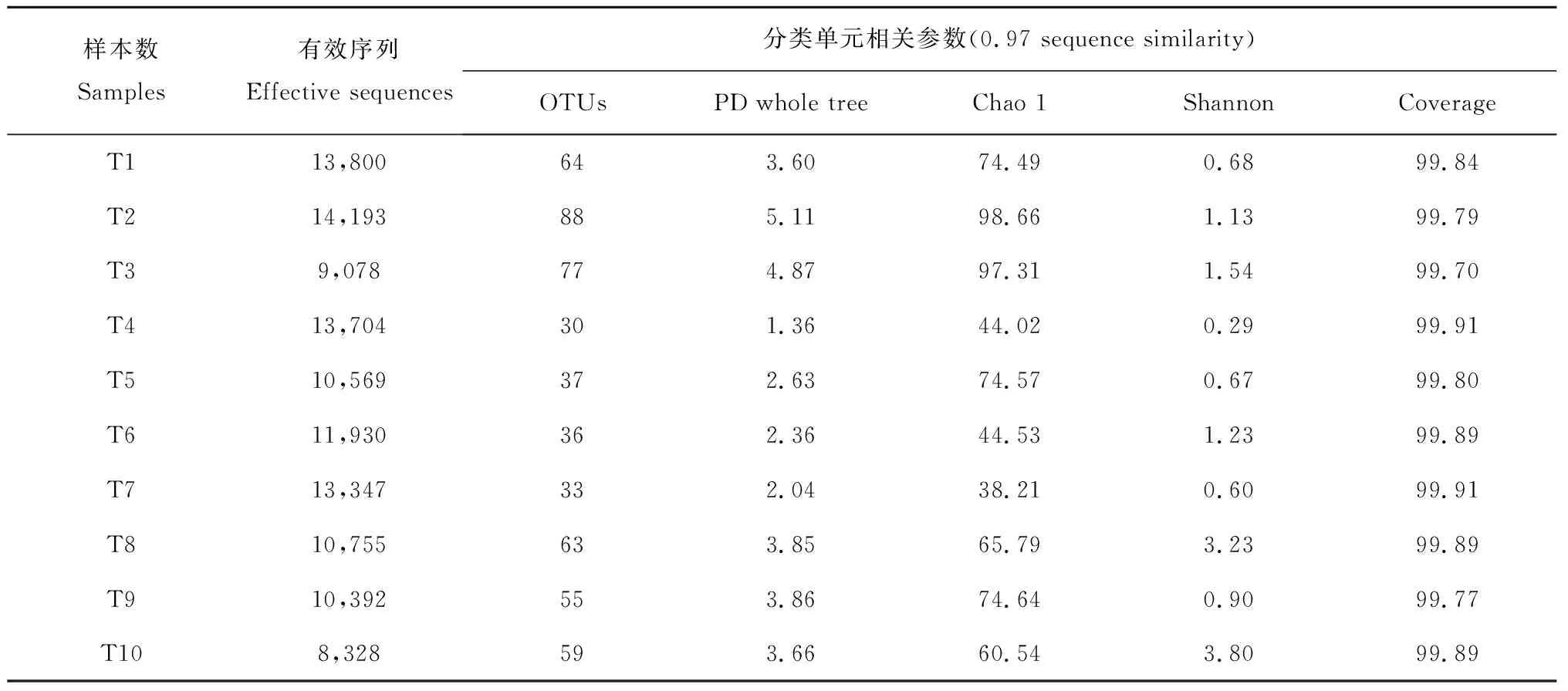
Table 1 Sequencing statistics and diversity analysis of oral microorganisms in the Tu nationality
2.2 群落结构与物种丰度分析
通过在RDP数据库进行相似性比对(156个细菌OTUs),获得分类地位明确的细菌共计7门12纲19目29科48属57种,以及大量未确定分类地位(Unclassified)的细菌。分别在属层次上对上述各样品的物种丰度进行统计,有关结果显示于图1。分析表明,土族牙周病主要的优势属群(丰度≥1%)是假单胞菌属Pseudomonas、魏斯氏菌属Weissella、嗜冷杆菌属Psychrobacter、芽孢杆菌属Bacillus、乳球菌属Lactococcus、克雷伯菌属Klebsiella、不动杆菌属Acinetobacter、肠杆菌科未知属Enterobacteriaceae unclassified、肠杆菌属Enterobacter、普罗维登斯菌属Providencia、肉食杆菌属Carnobacterium和明串珠菌属Leuconostoc。以上分析显示,牙周病患者口腔内微生物群落组成差异较大、种群结构复杂,优势类群在不同样本中各有差异。
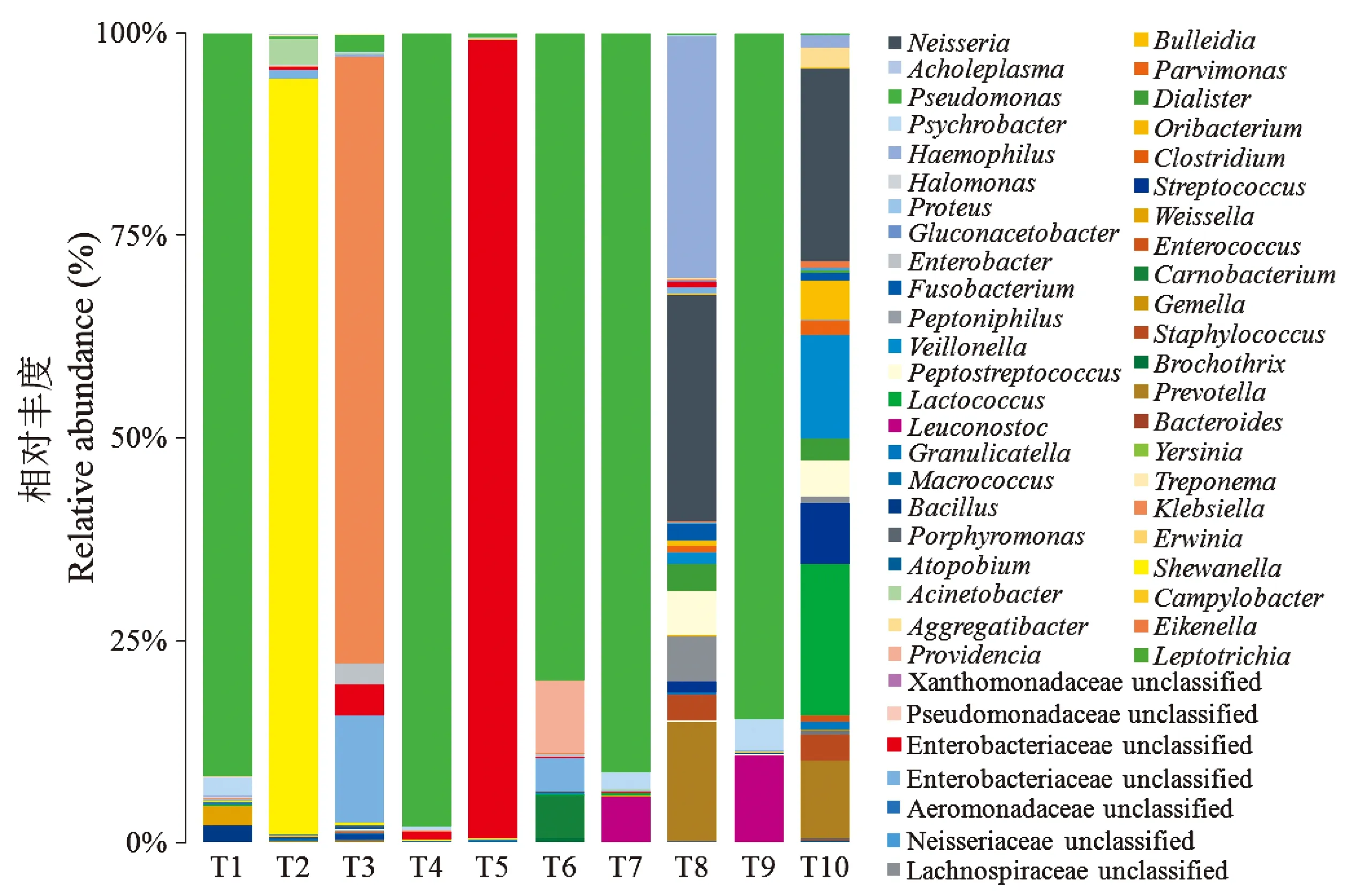
图1青海土族牙周病组微生物群落结构组成图
Figure1MicrobialcommunitystructureoftheperiodontaldiseasegroupsintheTunationalityfromQinghai
2.3 致病微生物分析
统计土族牙周病患者口腔内所有检出微生物的相对丰度,结果见表2。所检已知牙周病主要致病微生物种属(丰度比例)有消化链球菌属Peptostreptococcus(0.20%~5.50%)、微单胞菌属Parvimonas(0.80%~1.90%)、卟啉单胞菌属Porphyromonas(0.10%~0.20%)、普雷沃菌属Prevotella(0.10%~14.80%)、拟杆菌属Bacteroides(≤0.20%)、密螺旋体属Treponema(0.10%~0.20%)和小类杆菌属Dialister(2.80%~3.30%)。此外,还有大量可疑的致病微生物亦被检出,按丰度依次为(≥0.5%)肠杆菌科未知属Enterobacteriaceae unclassified、希瓦氏菌属Shewanella、克雷伯氏菌Klebsiella、奈瑟氏菌属Neisseria、嗜血杆菌属Haemophilus、乳球菌属Lactococcus、明串珠菌属Leuconostoc、韦荣球菌属Veillonella、普罗维登斯菌属Providencia、链球菌属Streptococcus、毛螺旋菌科未知属Lachnospiraceae unclassified、葡萄球菌属Staphylococcus、布雷德菌属Bulleidia和肉食杆菌属Carnobacterium。
表2土族牙周病组种群差异统计表
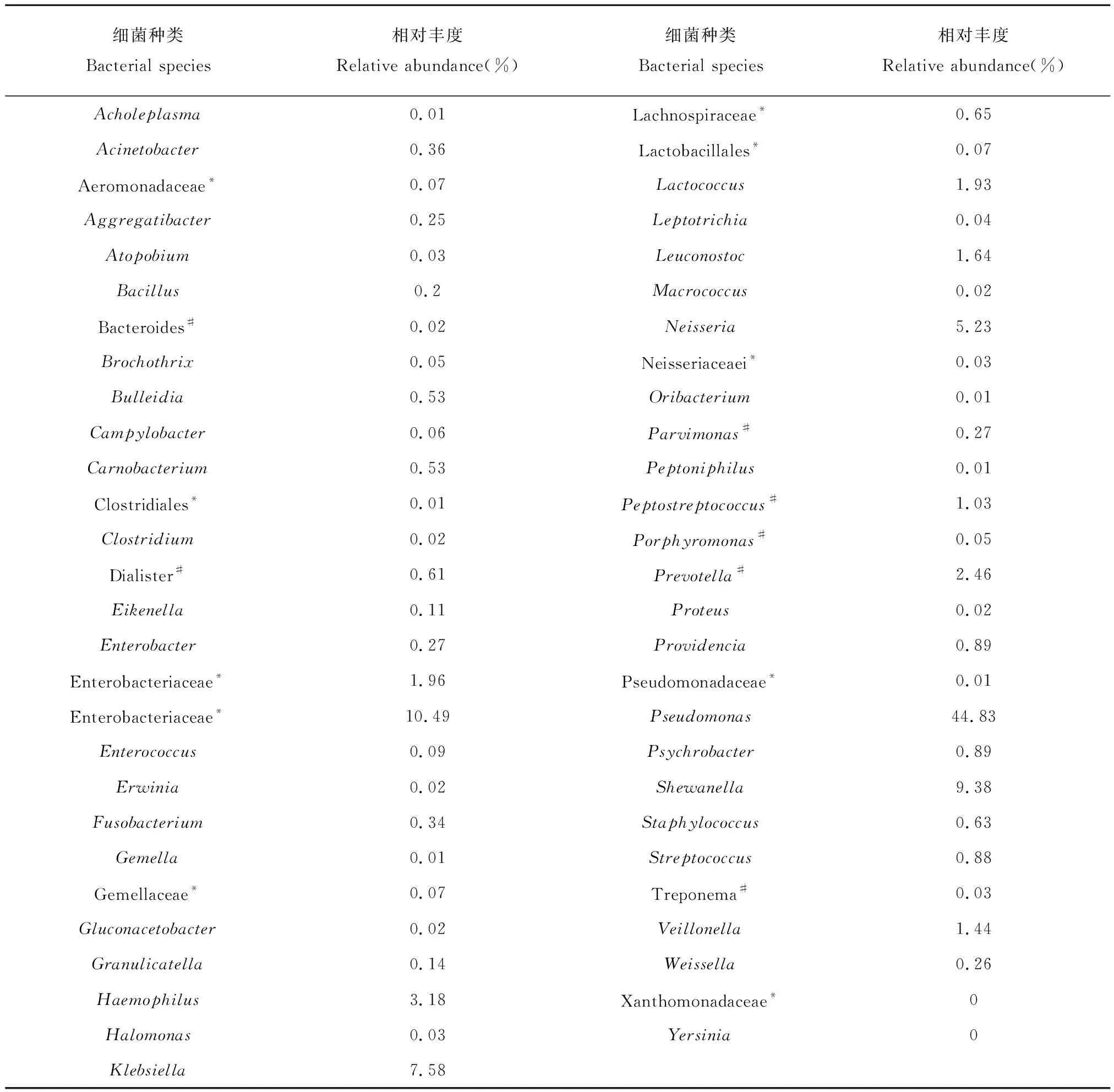
Table 2 Species statistics of periodontal disease group in Tu nationality
*:分类学地位尚不明确的属;0:表示未检出;#:文献报道的已知致病微生物.
3 讨论
目前,高通量测序是微生物多样性研究的重要技术手段,微生物多样性测序基于第二代高通量测序技术(Roche 454/MiSeq/HiSeq高通量测序),测序精确,数据准确度高,可全面反映复杂样品的微生物群落组成,以此可了解微生物群落与龋病、牙周病的相关性[8-9]。唾液样本由于能够提供整个口腔微生物群落信息,已作为诊断人体健康和口腔疾病的一种生物标记物[10-11]。本研究获得土族牙周病患者口腔微生物的物种注释OTU数目为156个,隶属于7门12纲19目29科48属57种,主要是放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、梭杆菌门(Fusobacteria)、螺旋体门(Spirochaetes)、无壁菌门(Tenericutes) 7个门,此与吴芳[12]等研究的东乡族牙周病人群相比,在OTU数量上有很大差异(60个OTU,共计6门27属),但Bacteroidetes、Fusobacteria、Firmicutes、Proteobacteria在两个民族牙周病人群中均有检出。同时,本研究发现牙周病患者口腔内的可观察物种和丰度指数Chao1、多样性指数Shannon及OTUs均高于或显著高于平原地区土族患者,显示不同海拔地区牙周病患者口腔内具有复杂的微生物群落结构,且是多种微生物(每个样本>30种细菌)共存互作的结果。
口腔微生物常以群体的方式引起各种口腔疾病,群体中的每一个个体均参与了疾病的发生发展过程,即使是低丰度的成员也可能作为复杂群落行为主要表现的关键物种[13]。比较分析土族牙周病的微生物种群差异,显示土族牙周病患者口腔内的致病微生物种类繁多,同时与其他民族比,可疑性致病微生物较多(57种)。牙周炎是由牙菌斑中多种微生物引发的牙周组织慢性感染性炎症疾病,其中卟啉单胞菌属(Porphyromonas)、密螺旋体属(Treponema)、坦纳菌属(Tannerella)、拟杆菌属(Bacteroides)、真杆菌属(Eubacterium)、消化链球菌属(Peptostreptococcus)、普氏菌属(Prevotella)、Parvimonasmicra、小类杆菌属(Dialister)、产线菌属(Filifactor)、脱硫球菌属(Desulfobulbus)及互养菌属(Synergistes)等与该病密切相关[14]。本研究中,已检测出明确的致病微生物包括Peptostreptococcus、Parvimonas、Porphyromonas、Prevotella、Bacteroides、Treponema和Dialister。此外,还有许多可疑的致病微生物亦被检出,如Shewanella、Klebsiella、Neisseria、Haemophilus、Lactococcus、Leuconostoc、Veillonella、Bulleidia和Carnobacterium等。
青海土族牙周病患者口腔内的微生物群落结构和可疑致病微生物类群,与已有诸多报道不一致,可能由青海地区环境、气候特征和饮食习惯差异造成。青海地处高海拔地区,盐碱化环境是青海地区的主要特征,饮食奶制品和牛羊肉较多,这可能是牙周疾病高发的主要原因。本研究还检出青海高原地区口腔特征微生物类群,如与乳酸发酵有关的乳杆菌属Lactobacillus、乳球菌属Lactococcus和明串珠菌属Leuconostoc等,这可能和青海地区人群的饮食生活习惯有关;与地区盐碱化环境有关的盐单胞菌属Halomonas以及与高原寒冷环境有关的Psychrobacter,该菌为革兰阴性球杆菌,因嗜冷而得名,属于莫拉菌科,广泛存在于海洋和陆地环境,能够在5 ℃下生长,一般在35 ℃~37 ℃范围内不能生长,这可能是青海特殊环境形成的特有菌群[15]。
本研究显示,青海土族牙周病患者口腔内Pseudomonas的含量较高,Pseudomonas多分布于土壤、淡水、海洋和生物体中,有极强分解有机物的能力,可以将多种有机物作为能量来源,生长温度范围较广,可在4 ℃~43 ℃范围内生长,大多数菌的最适温度在30 ℃左右,生长pH范围为7.0~8.5,且大部分属于耐冷菌,这可能与青海地区特殊的低温环境有关,具体的机制还需更深的研究[16]。
综上所述,本研究初步揭示了青海土族牙周病患者口腔内的微生物组成特征及种群结构的复杂多样性。土族牙周病的致病原因不仅有已知的牙周致病菌广泛参与,还可能存在大量可疑致病菌的协同参与。
