RP-HPLC法测定淫羊藿根中朝藿定C的含量
2018-09-21
黔南州食品药品检验所,贵州 都匀 558000
淫羊藿根为小檗科植物箭叶淫羊藿Epimediumsagittatum(Sieb. Et Zucc.) Maxim、巫山淫羊藿EpimediumwushaneneseT.S.Ying、天平山淫羊藿EpimediummyrianthumSteam、柔毛淫羊藿EpimediumpubescensMaxim、毡毛淫羊藿EpimediumcoactumH.R.Liang et W.M.Yan、粗毛淫羊藿EpimediumacuminatumFranch及光叶淫羊藿Epimediumsagittatumvar. glabratum T.S.Ying的干燥根。具有补肾壮阳、祛风除湿的功效,用于肾虚阳痿、小便淋漓、咳喘、风湿痹痛[1]。淫羊藿含有多种有效成分,其主要有效成分为淫羊藿苷类黄酮化合物[2],而其中的朝藿定C具有抗骨质疏松、抗癌、免疫增加等广泛的药理活性[3-4]。课题前期进行的预实验结果显示,朝藿定C的含量远高于淫羊藿苷等其他类黄酮化合物,适宜定量分析。故选择朝藿定C作为定量指标用于淫羊藿根药材的质量控制。本文用反相液相色谱测定了11种贵州地产药材淫羊藿根中朝藿定C的含量,可为淫羊藿根质量控制及相关医药产业提供理论根据参考。
1 仪器与材料
1.1 仪器 美国安捷伦1260高效液相色谱仪,上海科导SK250LHC超声波清洗器,瑞士梅特勒托利多AG135和AL204电子天平,北京普析TU-1901紫外分光光度计。
1.2 材料 朝藿定C对照品(含量95.5%,批号:111780-201503,中国药品生物制品检定所);乙腈(色谱纯,批号:20170403,天津科密欧);95%乙醇(分析纯,批号:20161105,天津科密欧);纯净水(批号:20171201,娃哈哈);淫羊藿根样品分别采集自贵州各地共11批(采集信息见表1),样品均经贵阳中医学院何顺志教授鉴定。样品洗净后晒干,粉碎,过三号筛,即得供试样品,置于干燥器中保存。
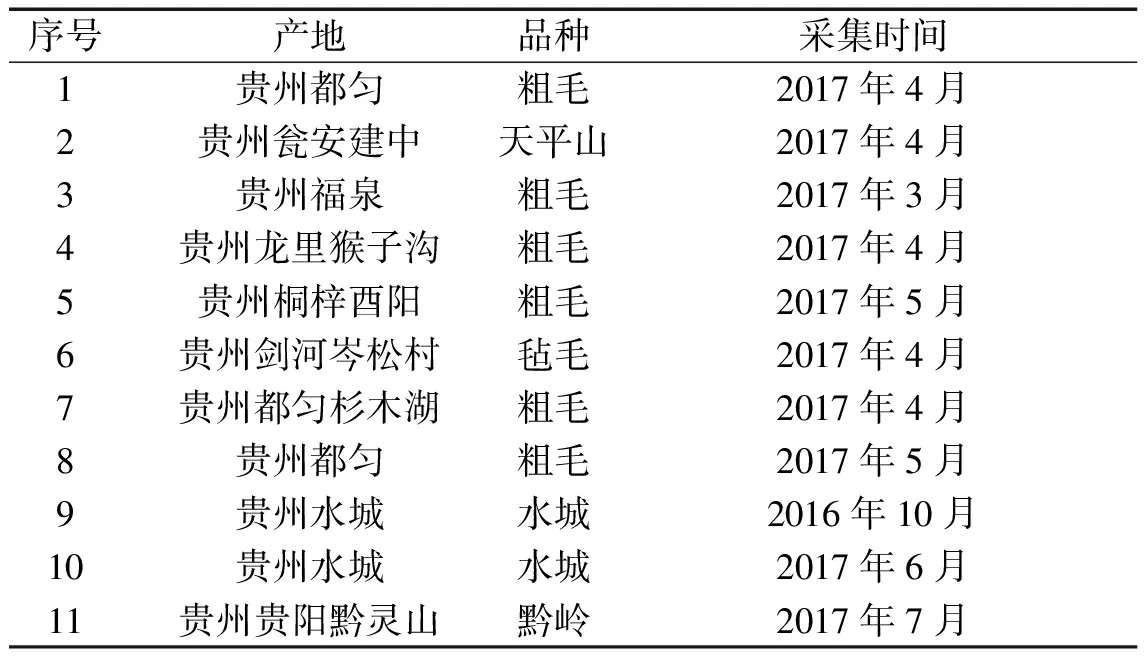
表1 淫羊藿根样品采集信息
2 方法与结果
2.1 色谱条件[5-6]色谱柱:美国安捷伦Eclipse Plus C18色谱柱(4.6 mm×250 mm,5 μm);流动相:乙腈-水(25∶75);流速:1.0 mL/min,进样量:10 μL;柱温:30℃;检测波长270 nm。理论板数以朝藿定C峰计算均不低于6000。
2.2 对照品溶液配制 精密称取朝藿定C对照品50.94 mg置50 mL量瓶中,加50%乙醇超声溶解并稀释至刻度,作为对照品贮备液;精密量取对照品贮备液5 mL置20 mL量瓶中,加50%乙醇稀释至刻度,即得(0.243 mg/mL)。
2.3 供试品溶液配制[5-6]取供试样品约0.2 g,精密称定,置具塞锥形瓶中,精密加入50%乙醇20 mL,称定重量,放置过夜,超声60分钟(50 kHz),放冷,再称定重量,用50%乙醇补足减失的重量,摇匀,经0.45 μm滤膜滤过,取续滤液,即得。
2.4 峰纯度考察 取对照品溶液、供试品溶液各10 μL,分别注入液相色谱仪,按2.1项下的色谱条件,依法测定。朝藿定C峰的紫外扫描光谱表明:朝藿定C对照品的紫外扫描图在200 nm、270 nm和320 nm处有最大吸收。3D图谱显示:200 nm波长处吸收大但杂质干扰较多,320 nm波长处干扰少但吸收较小,270 nm波长处吸收值较大且干扰少。供试品溶液中朝藿定C峰的紫外扫描图与对照品峰的紫外扫描图谱一致,且纯度因子达到999以上。综合以上考察,确定检测波长为270 nm。
2.5 线性关系考察 精密量取2.2项下对照品贮备液1、2、4、6、8 mL分别置10 mL量瓶中,加50%乙醇稀释至刻度,得一系列浓度级别的对照品溶液;精密吸取上述系列对照品溶液和对照品贮备液各10 μL,分别注入液相色谱仪,按2.1项下的色谱条件测定,记录色谱图,以峰面积为纵坐标,浓度(mg/mL)为横坐标,进行线性回归计算,朝藿定C回归方程为:y=18219.1343x+26.8045,r=1.0000,结果表明朝藿定C在0.0973 ~0.9730 mg范围内呈良好的线性关系。
2.6 精密度考察 精密吸取2.2项下朝藿定C对照品溶液10 μL,按2.1项下的色谱条件,注入液相色谱仪,连续进样6次,测定峰面积,朝藿定C峰面积的RSD为0.14%,结果表明方法有良好的精密度。
2.7 稳定性考察 取同一供试品溶液分别于0、5、11、18 h进样,进样量10 μL,按2.1项下的色谱条件,测定朝藿定C峰面积,RSD为0.6%,表明供试品溶液在18 h内稳定性良好。
2.8 重复性考察 称取同一供试品6份,按2.3项下方法配制供试品溶液,按2.1项下的色谱条件,进样量10 μL,测定峰面积,计算含量。供试品中朝藿定C的平均含量为2.611%,RSD为0.4%,表明本方法重复性良好,符合定量标准要求。
2.9 加样回收率试验 精密称取8份已测定含量的样品0.1 g(朝藿定C含量为2.611%),分别置具塞锥形瓶中,精密加入回收对照溶液20 mL,称定重量,放置过夜,超声60 min,放冷,再称定重量,用50%乙醇补足减失的重量,摇匀,0.45 μm滤膜滤过,取续滤液,按2.1项下的色谱条件,进样10 μL,记录色谱图,计算回收率。朝藿定C的平均回收率为102.3%,RSD为0.7%,表明本方法准确度良好,符合相关规定。结果见表2。
2.10 样品含量测定 取不同产地供试样品按2.3配制供试品溶液,分别进样10 μL,记录色谱图,按外标法以峰面积计算含量。结果见表3。

表2 淫羊藿根中朝藿定C的回收率实验结果

表3 淫羊藿根中朝藿定C含量测定结果 (%)
3 讨论
为获得准确的含量测定结果,本研究考察了浸泡过夜、浸泡过夜后超声、浸泡过夜后水浴回流、浸泡过夜后超声再水浴回流等四种不同的提取方法,结果表明,浸泡过夜后超声效果为最佳,增加水浴回流步骤反而使朝藿定C含量结果降低。文献报道[7],朝藿定C(三糖苷)在高温环境下会脱去一个鼠李糖后转化为淫羊藿苷(二糖苷),导致朝藿定C含量下降、淫羊藿苷含量升高。因此选择浸泡过夜后超声作为本实验的提取方式。
实验中有3个样本分别为黔岭淫羊藿和水城淫羊藿,不在贵州省淫羊藿根药材标准收载的品种中,但作为贵州特有品种,本研究也对这3个样本进行了含量测定,朝藿定C含量最低为0.86%,而标准收载的品种中,朝藿定C含量最低为0.89%。结果显示,非标准品种中朝藿定C的含量也可以达到标准品种的含量水平,建议修订淫羊藿根标准时增加收载品种,扩大贵州省淫羊藿根来源的覆盖范围。 淫羊藿根作为贵州省少数民族常用药材,收载于《贵州省中药材民族药材质量标准》中,但标准中仅有一项性状描述,不能有效控制药材的质量。已有文献研究对象多为淫羊藿药材,按《中国药典》一部定义为淫羊藿的干燥叶[8]。淫羊藿植株入药部位不同,有效成份含量也有所差异,本研究可为淫羊藿根药材的标准制定、医疗应用和质量控制提供数据参考。
