螺旋霉素分子印迹磁性纳米吸附剂的合成及应用
2018-08-02孙佳佳章飞芳梁鑫淼
孙佳佳, 章飞芳, 梁鑫淼
(华东理工大学药学院, 上海 200237)
大环内酯类抗生素(macrolide antibiotics, MA)是一类弱碱性的亲脂性分子,广泛用于治疗人类和动物的细菌感染以及作为育种行业的生长促进剂。抗生素的滥用会导致食品中的抗生素残留,对人体产生不良影响[1]。为保证食物安全和消费者的健康,许多国家都对MA设置了最大残留限量要求(maximum residue limits, MRLs)[2]。比如欧盟对牛奶中替米考星、红霉素和螺旋霉素的最大残留限量分别为50、40和200 μg/kg[3]。
目前,MA的检测方法主要有毛细管电泳-电化学发光检测[4]、高效液相色谱-质谱(HPLC-MS)[5-8]、高效液相色谱(HPLC)结合蒸发光散射检测器(ELSD)[9]或紫外检测器(UVD)[10]。虽然HPLC-MS检测灵敏度高、适用范围广,但其价格极其昂贵,且日常维护成本高的缺陷限制了该方法的普及。由于多数HPLC均配置有UVD,利用该配置发展出相应的方法显然有助于方法的普及。由于多数MA缺少特定的生色基团或吸收系数不高,导致方法检出限较高,无法用于痕量目标物的测定。有必要通过前处理对目标抗生素进行有效富集。
目前用于处理MA样品的前处理技术主要是固相萃取(solid phase extraction, SPE),包括其衍生技术,例如分散固相萃取[7]、磁性固相萃取[10]、固相微萃取[11]。在众多SPE吸附剂中,分子印迹聚合物(molecularly imprinted polymers, MIPs)因为能对目标分析物或其结构类似物进行特异性吸附而受到关注。纳米尺寸的MIPs比传统块状聚合物具有更多优点,比如模板分子容易去除、传质阻力小且结合速度快[12]。磁性核壳结构分子印迹纳米粒子由一个磁性核和分子印迹聚合物壳组成,通过表面分子印迹技术使结合位点位于材料表面,有助于提高萃取效率[13],并且内部的磁性核还可以在外加磁场作用下实现相分离,消除了传统SPE方法冗长的处理过程[14],同时可以使用少量吸附剂处理大体积样品,有利于得到更高的富集倍数。Pérez等[15]合成油酸盐功能化的磁性纳米粒子,结合LC-MS/MS测定不同来源水样中的3种MA。Xie等[9]以红霉素、四环素及氯霉素为混合模板分子,制备出相应的MIP用于SPE吸附剂,采用HPLC-ELSD对牛奶中的红霉素、四环素和氯霉素进行分析。Zheng等[16]以泰勒菌素为虚拟模板,制备出相应的MIP用作SPE吸附剂,建立HPLC-UV方法检测5种饲料中的替米考星。Song等[17]以泰拉霉素为模板分子,通过沉淀聚合法合成相应的MIP,以分散固相萃取的方式结合LC-MS/MS检测,同时测定猪肉中的7种MA残留。这些吸附剂的共同特点是无磁性。因此在萃取后,与溶液的分离仍然依靠过滤方式实现。目前,采用MA为模板分子的分子印迹磁性纳米吸附剂鲜有报道。
本文以磁性纳米Fe3O4为内核,合成出以螺旋霉素(spiramycin, SPI)为模板分子的核壳型分子印迹磁性纳米吸附剂,优化其合成条件并将其用于蜂蜜样品中MA的检测。
1 实验部分
1.1 材料和试剂
4种MA标准品包括螺旋霉素(SPI,纯度97% )、交沙霉素(josamycin, JOS,纯度91.2% )、替米考星(tilmicosin, TIL,纯度99% )和酒石酸泰勒菌素(tylosin tartrate, TYL,纯度98.9% )(德国Dr. Ehrenstorfer公司);丙烯酸(acrylic acid, AA)、甲基丙烯酸(methacrylic acid, MAA)和乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate, EDMA)(色谱纯,上海阿拉丁试剂有限公司);偶氮二异丁腈(2,2′-azobis(2-methylpropionitrile), AIBN,色谱纯,百灵威科技有限公司);二甲基亚砜(dimethyl sulfoxide, DMSO,分析纯,上海麦克林生物科技有限公司);冰乙酸(分析纯,上海天莲化工科技有限公司);聚乙烯吡咯烷酮(polyvinyl pyrrolidone, PVP,优级纯,国药集团化学试剂有限公司);甲醇(色谱纯,美国TEDIA公司); Fe3O4颗粒(纯度99.5% ,粒径20 nm,阿达马斯试剂有限公司);甲醇、无水乙醇、氯化钠、磷酸、磷酸氢二钠和磷酸二氢钠(分析纯,上海凌峰化学试剂有限公司)。
1.2 色谱仪器
岛津LC-20A高效液相色谱仪,包括LC-20AD输液泵、DGU-20A3R在线脱气机、SIL-20AC自动进样器、SPD-20A检测器、LCsolution色谱工作站;XCharge C18色谱柱(150 mm×4.6 mm, 3.5 μm)(华谱新创科技有限公司); Milli-Q超纯水发生器(美国Millipore公司)提供配制溶液的纯水。
1.3 螺旋霉素-分子印迹磁性纳米吸附剂的合成
在500 mL圆底烧瓶中加入1 g Fe3O4和200 mL无水乙醇,超声5 min使其分散均匀,之后加入5 mL AA;混合物在35 ℃、300 r/min的机械搅拌下反应2 h,得到Fe3O4@AA。用去离子水和无水乙醇各清洗3遍,之后置于60 ℃烘箱中干燥12 h备用。
在100 mL圆底烧瓶中加入SPI(0.125 mmol, 105.38 mg), MAA(1.75 mmol, 150 μL)和无水DMSO(40 mL),在30 ℃、500 r/min转速下磁力搅拌1 h,进行预聚合。然后加入250 mg上述处理得到的Fe3O4@AA,以及EDMA(3.5 mmol, 660 μL)和PVP(100 mg);超声5 min使混合均匀,然后加入60 mg AIBN;在N2保护条件下,60 ℃、700 r/min转速下磁力搅拌24 h,进行自由基聚合反应。磁场分离得到固体颗粒,去离子水和乙醇各清洗3次,得到最终的吸附剂Fe3O4@AA@SPI@MIP。其合成路线如图1所示。
非印迹聚合物Fe3O4@AA@NIP的合成步骤与上述过程一样,区别在于无模板分子加入。
使用40 mL 20% (v/v)乙酸-甲醇超声30 min处理上述Fe3O4@AA@SPI@MIP,重复洗脱3次,取每次洗脱液各7 mL,氮气吹干后用10% (v/v)甲醇-水重溶至500 μL。经HPLC-UV检测,后两次洗脱液中都没有SPI的紫外吸收,证明模板分子去除干净。最终将吸附剂在60 ℃烘箱中干燥12 h。
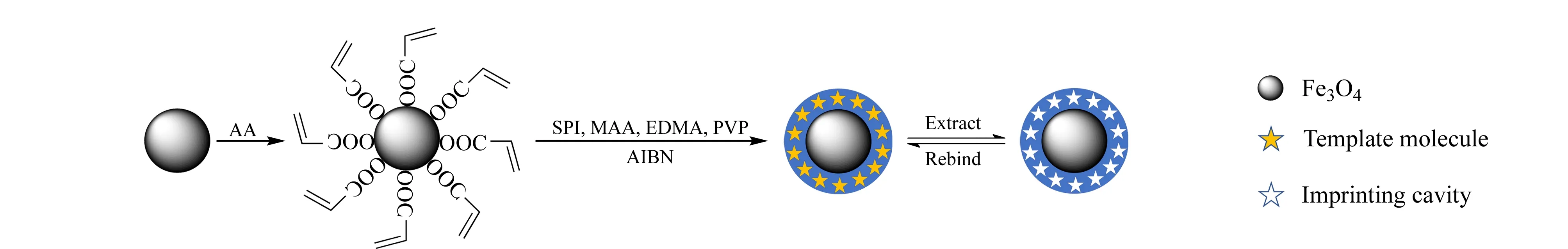
图 1 Fe3O4@AA@SPI@MIP合成路线示意图Fig. 1 Schematic diagram of synthesis route of Fe3O4@AA@SPI@MIP
1.4 Fe3O4@AA@SPI@MIP吸附实验
饱和吸附量的考察:将10 mg Fe3O4@AA@SPI@MIP和Fe3O4@AA@NIP分别分散在50 mL锥形瓶中。每个锥形瓶中加入20 mL不同质量浓度的SPI水溶液(0.5~20 μg/mL),超声萃取10 min后,在30 ℃恒温条件下放置1 h进行吸附,之后通过磁场相分离。用水简单淋洗吸附剂表面,再用8 mL 20% (v/v)乙酸-甲醇超声10 min,使SPI从吸附剂中完全洗脱。洗脱液在40 ℃用氮气吹干后用10% (v/v)甲醇-水重溶至500 μL,经0.22 μm有机膜过滤后检测。根据公式(1)计算不同质量浓度下达到吸附平衡的吸附量(Qe, mg/g):
Qe=Ce×V/M
(1)
其中,Ce(μg/mL)是达到吸附平衡时目标分析物洗脱重溶后的质量浓度,V(mL)是重溶体积,M(mg)是吸附剂的质量。
吸附平衡时间的考察:同样取10 mg上述两种吸附剂,使用质量浓度为1.5 μg/mL的SPI水溶液上样。萃取过程同上。在30 ℃恒温条件下放置10~120 min。根据公式(2)计算不同时间的吸附量(Qt, mg/g):
Qt=Ct×V/M
(2)
其中,Ct(μg/mL)是不同吸附时间下目标分析物洗脱重溶后的质量浓度。
Fe3O4@AA@SPI@MIP的识别能力采用印迹因子α表征,参考相关文献[18,19],印迹因子的计算公式为:
α=QMIP/QNIP
(3)
其中,QMIP和QNIP(mg/g)分别为Fe3O4@AA@SPI@MIP和Fe3O4@AA@NIP对SPI的饱和吸附量。
1.5 蜂蜜样品的前处理
在空白蜂蜜样品中添加10~200 μg/L(0.1~2 mg/kg)的4种MA标准品,绘制基质添加标准曲线;在低中高3个水平(50、100和150 μg/L)计算加标回收率和精密度。
2 结果和讨论
2.1 Fe3O4@AA@SPI@MIP合成条件的优化
Fe3O4的功能化通过丙烯酸的羧基和磁性纳米铁表面残留的Fe3+发生络合反应进行[20]。合成条件的优化结果(数据未提供)表明,无水DMSO作为致孔溶剂效果好,模板分子/功能单体/交联剂的物质的量之比为1∶14∶28时效果最佳,Fe3O4@AA与反应单体(包括模板分子、功能单体和交联剂)的比例为250 mg∶0.125 mmol(SPI)时吸附性能最好。
2.2 吸附剂的表征
Fe3O4@AA@SPI@MIP的BET表征结果显示,Fe3O4@AA@SPI@MIP比表面积为11.63 m3/g,略大于Fe3O4@AA@NIP的比表面积(8.70 m3/g)。由扫描电镜(SEM)表征结果看出,吸附剂基本呈球形(见图2), Fe3O4的粒径为20 nm, Fe3O4@AA@SPI@MIP的粒径增至约200 nm, Fe3O4@AA@NIP的粒径约为150 nm。在反应单体用量相同的情况下,合成MIP过程中加入了模板分子,使得聚合物表面及壳层中都存在许多印迹位点,相比NIP的聚合层增加了印迹空腔的空间,所以MIP具有更大的粒径。去除模板分子后的MIP留有印迹空腔,具有增大比表面积的作用。

图 2 (a)Fe3O4@AA@SPI@MIP和(b)Fe3O4@AA@NIP 的扫描电镜图Fig. 2 SEM images of (a) Fe3O4@AA@SPI@MIP and (b)Fe3O4@AA@NIP
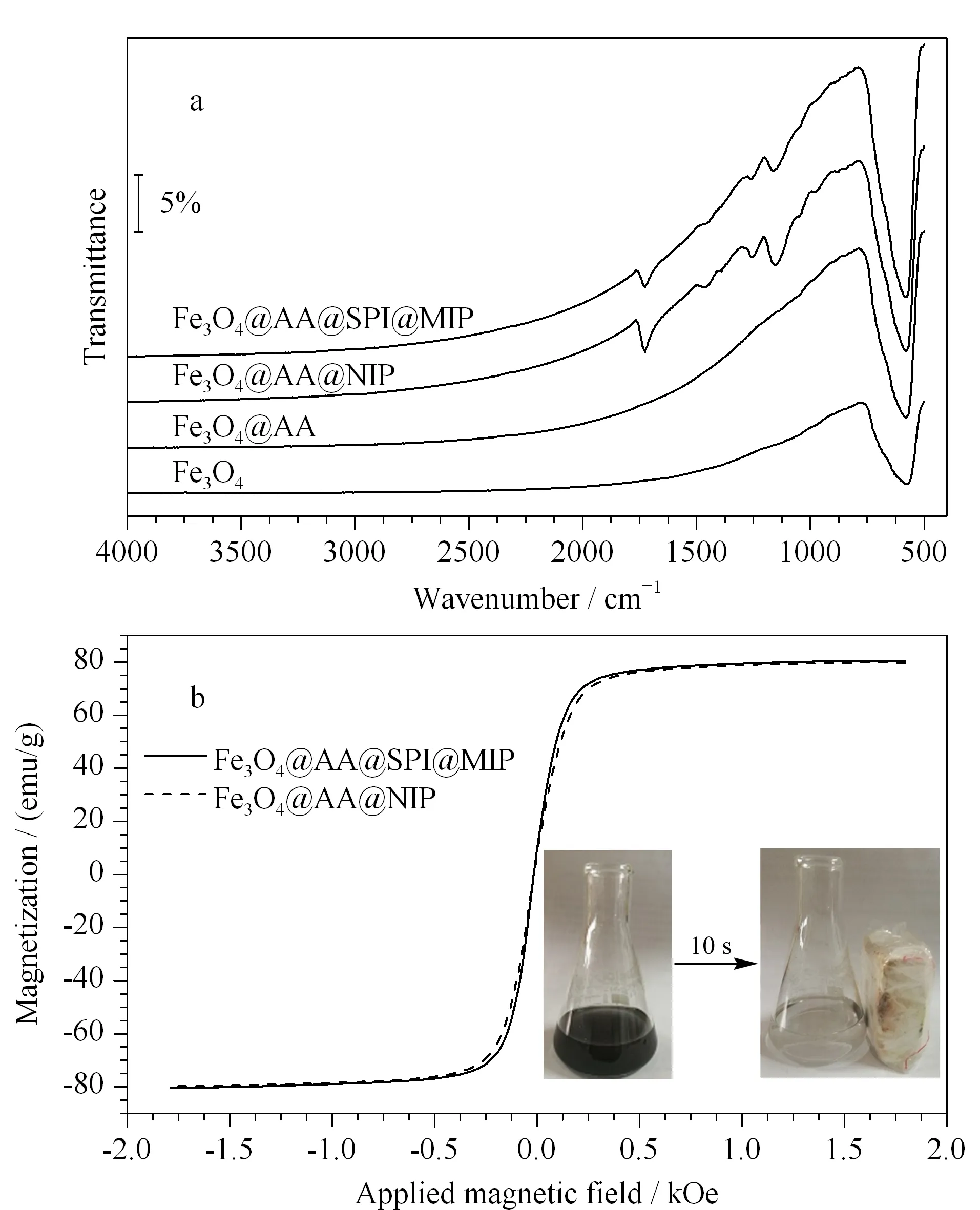
图 3 磁性纳米粒子的(a)FT-IR和(b)VSM图Fig. 3 (a) FTIR and (b) VSM spectra of magnetic nanoparticles
FT-IR及振动样品磁强计谱(VSM)的表征结果见图3。Fe3O4@AA@SPI@MIP和Fe3O4@AA@NIP在1 161 cm-1和1 730 cm-1处有特征吸收,分别对应饱和酯类的-C-O和-C=O的伸缩振动,表明在磁铁表面成功进行了聚合反应。需要指出的是,低波数处的基线有漂移,这可能是与样品自身特有性质有关(样品呈黑色,是按照样品和KBr质量比为1∶100压制得到的薄片),但这并不影响定性分析;Fe3O4@AA在578 cm-1处的振动吸收增强,可能是配位键的存在使得Fe-O键不对称性增强,振动吸收增强。根据测量,Fe3O4@AA@SPI@MIP的磁化强度为80.408 emu/g, Fe3O4@AA@NIP的磁化强度为79.834 emu/g,两者结果接近。据文献[21]报道,粒径16 nm的商品化Fe3O4磁化强度为84.5 emu/g。所以,最终得到的吸附剂磁性并未受到聚合层的影响,在外加磁场下能够实现快速分离(见图3)。文献报道以罗丹明B羟脯氨酸衍生物为模板分子的磁性MIP的磁化强度为20.6 emu/g[22],以吗啡为模板分子的磁性MIP的磁化强度为13.02 emu/g[23]。相比之下,本文的吸附剂具有更高的磁化强度,高磁化强度显然更利于萃取后的有效相分离。
2.3 吸附实验结果
吸附平衡曲线和动力学吸附曲线见图4。由图4a可以看出,在低浓度区两种吸附剂的吸附量很接近;随着样品浓度的增加,Fe3O4@AA@SPI@MIP的吸附量增加幅度明显高于Fe3O4@AA@NIP,相应的二者吸附量差异明显变大。当样品质量浓度达到16 μg/mL时,Fe3O4@AA@SPI@MIP的吸附趋于饱和。通过计算可得到Fe3O4@AA@SPI@MIP的饱和吸附量QMIP=2.37 mg/g,远高于Fe3O4@AA@NIP的饱和吸附量(QNIP=0.67 mg/g)。这显然有利于得到更高的吸附容量。另外,Fe3O4@AA@SPI@MIP的印迹因子α为3.54;在1.5 μg/mL下,两种吸附剂随着静置时间的增加,吸附量增加,60 min达到吸附平衡。
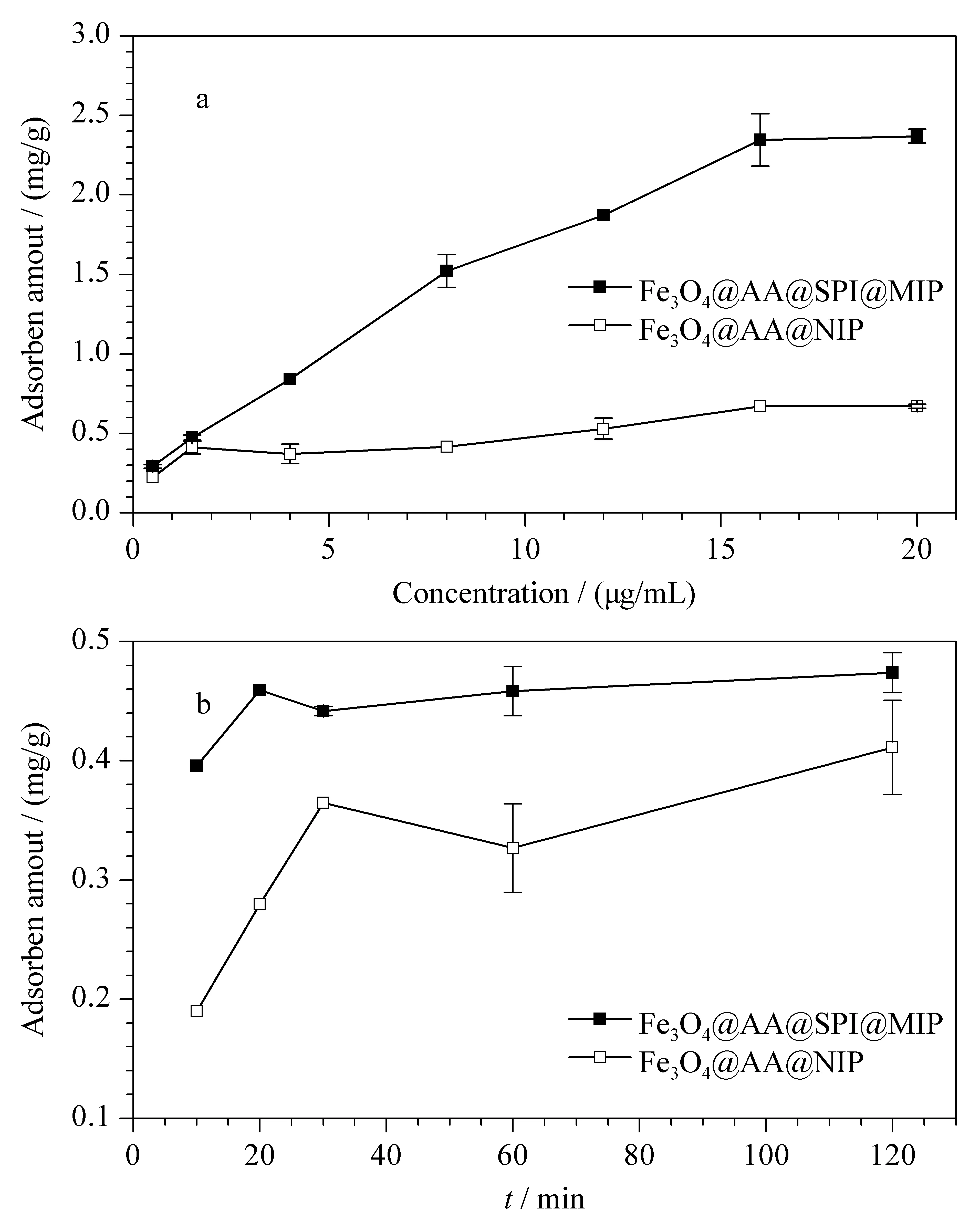
图 4 Fe3O4@AA@SPI@MIP和Fe3O4@AA@NIP对螺旋 霉素的(a)吸附平衡曲线和(b)动力学吸附曲线(n=3)Fig. 4 (a) Adsorption equilibrium curve and(b) kinetic binding curve of spiramycin toward Fe3O4@AA@SPI@MIP and Fe3O4@AA@NIP (n=3)
2.4 富集效果考察
Fe3O4@AA@SPI@MIP对4种MA的富集效果见图5。与直接进样对比,4种MA(SPI、JOS、TIL、TYL)通过吸附剂处理之后均得到了有效的富集。按照文献[10]的计算方法,上述4种MA富集倍数分别为310、118、758和72。该吸附剂对TIL具有最高的富集倍数。可能是因为TIL和SPI化学结构相似,且空间结构小于SPI,因而更容易被印迹位点识别并结合。
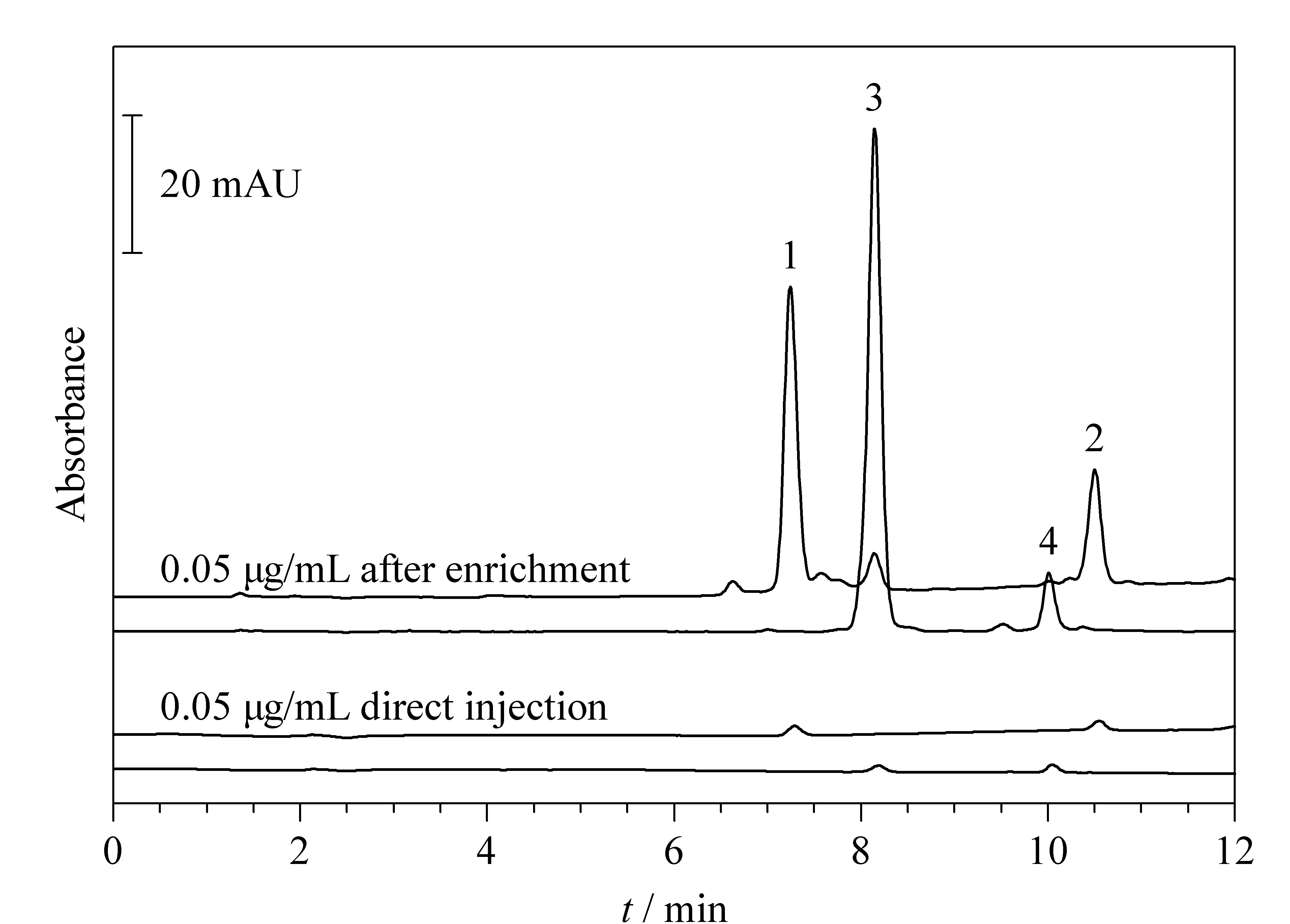
图 5 Fe3O4@AA@SPI@MIP对4种MA的富集效果图Fig. 5 Enrichment effect of Fe3O4@AA@SPI@MIP towards four MA Mobile phase: A, 0.02% (v/v) H3PO4/H2O; B, MeOH. Gradient mode: 0-10 min, 10%B-90%B; 10-12 min, 90%B. UV detection: 232 nm (SPI, JOS) and 282 nm (TIL, TYL); injection volume: 20 μL; flow rate: 1 mL/min; column temperature: 25 ℃; loading volume: 200 mL; redissolution volume: 200 μL. 1. spiramycin; 2. josamycin; 3, tilmicosin; 4. tylosin tartrate.
2.5 萃取条件优化和循环使用次数
为保证Fe3O4@AA@SPI@MIP达到最佳萃取效果,对其萃取条件进行了优化(数据未显示)。结果表明,当上样溶液pH近乎中性(pH 6)、不含无机盐时可达到最佳吸附效果,洗脱溶液为5% (v/v)乙酸-甲醇、洗脱液体积为3 mL即可达到目标抗生素的完全洗脱。将上述优化的萃取条件用于后续蜂蜜样品的萃取实验。优化结果的可能原因是,目标抗生素在甲醇中易溶,而乙酸的存在会破坏目标抗生素与吸附剂间的氢键作用。弱碱性的抗生素在弱酸性溶液中带正电荷,与Fe3O4@AA@SPI@MIP结构中的羧基具有离子间作用,和氢键作用共同影响吸附剂和抗生素间的相互作用,在pH 6时达到最佳吸附效果。无机盐的存在会干扰目标分子和吸附剂间的离子键作用及氢键作用。在优化后的萃取条件下,取3份Fe3O4@AA@SPI@MIP,每份10 mg,重复使用6次。结果表明,6次重复得到的峰面积基本保持不变(所得峰面积相对标准偏差为5% ~11% ),这表明该吸附剂可重复使用。
2.6 蜂蜜样品中MA的萃取和定量
方法的线性范围、线性方程、相关系数、检出限及定量限结果见表1。蜂蜜样品加标回收率和精密度结果见表2。结果显示,在10~200 μg/L(0.1~2 mg/kg)质量浓度范围内,4种MA的峰面积(Y)和对应的质量浓度(X, μg/L)的线性方程呈现出良好的线性关系(R2≥0.99); 4种MA的检出限为0.53~2.75 μg/L,定量限为1.78~9.16 μg/L;方法回收率在80.78% ~123.02%之间,相对标准偏差(RSD)<15.85% 。
Leal等[24]采用HPLC-UV测定鸡肉样品中几种抗生素。该方法对交沙霉素和SPI抗生素的检出限范围为6~33 μg/L。尽管由于基质不同无法直接类比,但仍可以看出本文方法具有较低的检出限。
表14种MA的线性范围、线性方程、相关系数、LOD以及LOQ(n=3)
Table1Linearranges,linearequations,correlationcoefficients(R2),LODandLOQofthefourmacrolideantibiotics(n=3)

AnalyteLinear range/(μg/L)Linear equationR2LOD/(μg/L)LOQ/(μg/L)Spiramycin10-200Y=(343.7±20.4)X+(5467.9±1264.1)0.98982.759.16Josamycin10-200Y=(615.2±31.9)X+(6436.2±1925.6)0.99511.906.34Tilmicosin10-200Y=(1064.1±7.9)X+(15190.5±449.8)0.99730.531.78Tylosin tartrate10-200Y=(209.5±15.2)X+(2484.4±859.7)0.99080.902.99
LOD and LOQ are calculated at signal to noise ratio of 3 and 10, respectively;Y: peak area;X: mass concentration, μg/L.
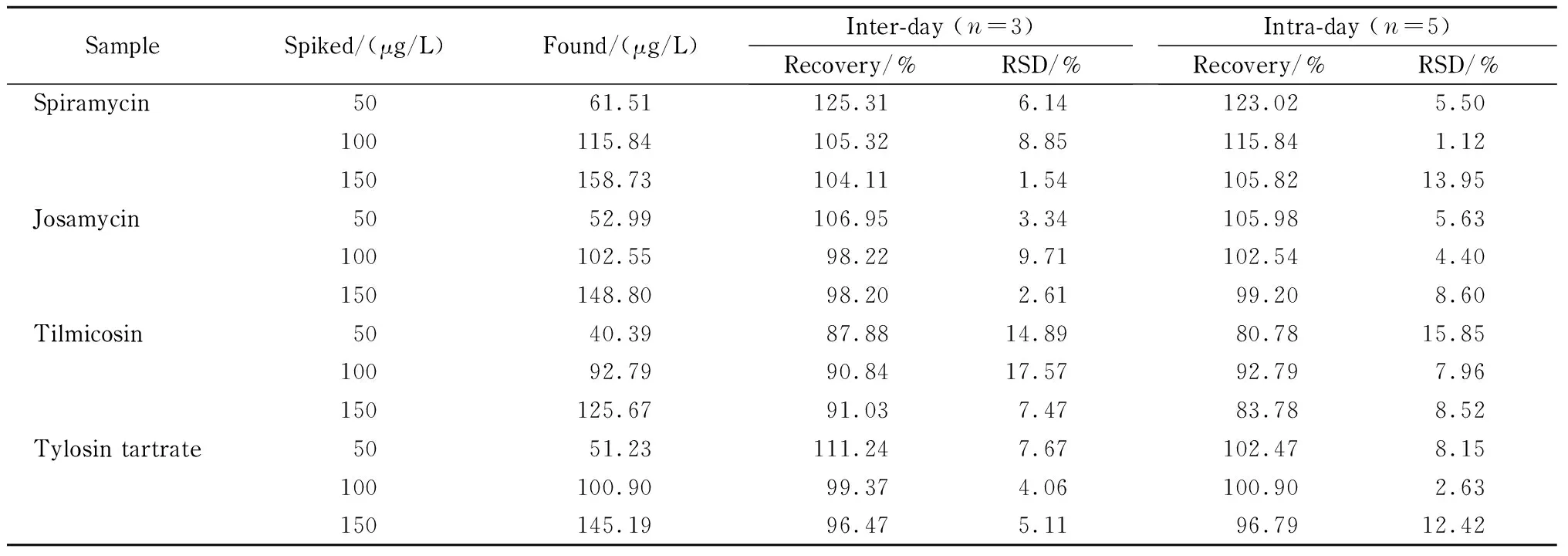
表 2 蜂蜜样品中3个水平下的MA加标回收率Table 2 Recoveries of macrolide antibiotics in honey samples spiked at three levels
Background: not detected.
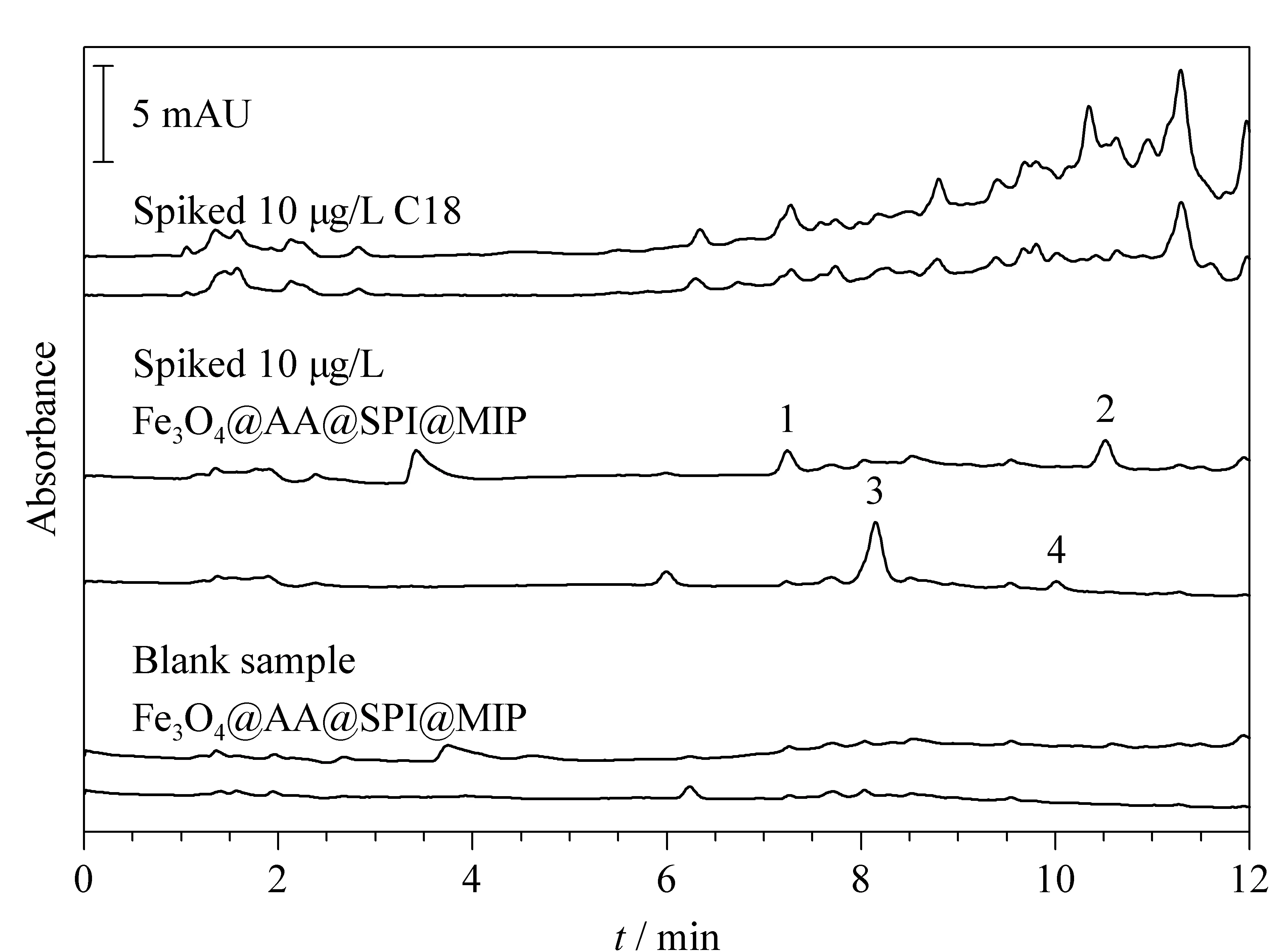
图 6 C18和Fe3O4@AA@SPI@MIP净化蜂蜜样品的效果对比Fig. 6 Comparison of purification effect on honey sample between C18 and Fe3O4@AA@SPI@MIP Conditions are the same as Fig. 5 except that the loading volume is 100 mL. 1. spiramycin; 2. josamycin; 3, tilmicosin; 4. tylosin tartrate.
另外,为了考察所发展吸附剂的选择性,特选择常规C18型吸附剂进行了对比,结果如图6所示,可以看出本文所发展的Fe3O4@AA@SPI@MIP吸附剂在处理蜂蜜样品时能有效净化基质,干扰物质少。相比之下C18不存在特异性吸附,基质干扰严重,无法对添加的痕量目标抗生素进行富集,故无法准确定量。
3 结论
本文合成出螺旋霉素为模板分子的分子印迹磁性纳米吸附剂。该吸附剂对MA表现出良好的富集效果,可结合HPLC-UV法用于蜂蜜样品中4种目标MA的测定。该吸附剂还有望用于处理含其他类型的MA,为HPLC-UV法测定包括蜂蜜在内的其他食品中痕量MA提供了一种样品前处理的新选择。
猜你喜欢
杂志排行

色谱的其它文章
- 全二维气相色谱-质谱分析煤油基吸热型碳氢燃料烃族组成
- QuEChERS-超高效液相色谱-串联质谱法测定不同植被类型土壤中11种甲氧基丙烯酸酯类杀菌剂
- 离子液体分散液液微萃取-超高效液相色谱-串联质谱法测定食品接触材料中全氟辛酸和全氟辛烷磺酸的迁移量
- 超高效液相色谱-串联质谱法快速测定鸡蛋中氟虫腈及其代谢物残留
- 固相萃取-高效液相色谱-串联质谱法同时测定牛奶和羊奶中莫奈太尔及其代谢产物残留
- Determination of 11 synthetic musks in imported seafood by solid phase extraction and gas chromatography-mass spectrometry