肝癌细胞HepG2中增强子的识别及生物信息学分析
2017-11-06丁若凡李宇鹏张一鸣朱小冬胡海碧刘文荣李玲郭志云
丁若凡,李宇鹏,张一鸣,朱小冬,胡海碧,刘文荣,李玲,郭志云
1.西南交通大学 生命科学与工程学院,四川 成都 610031;2.成都市第三人民医院 病理科,四川 成都 610031
肝癌细胞HepG2中增强子的识别及生物信息学分析
丁若凡1,李宇鹏1,张一鸣1,朱小冬1,胡海碧1,刘文荣1,李玲2,郭志云2
1.西南交通大学 生命科学与工程学院,四川 成都 610031;2.成都市第三人民医院 病理科,四川 成都 610031
目的:整合增强子特征识别肝癌细胞HepG2增强子,并对其保守性、GC含量、转录因子调控、靶基因功能等进行分析,以期解析肝癌细胞增强子参与的调控网络。方法:通过整合H3K27ac、H3K4me1和H3K4me3组蛋白修饰及DNaseⅠ高敏位点的Chip-seq数据预测HepG2中的增强子,计算每个增强子的平均PhastCons分数和GC含量,评估整体增强子的保守性与GC含量,整合ENCODE转录因子结合位点数据寻找转录因子-增强子调控,使用GREAT和DAVID分别对增强子和增强子的靶基因进行GO与KEGG通路功能富集分析。结果:共识别2254个肝细胞癌增强子,1432个增强子靶基因,135个转录因子的9983个增强子结合位点;比较随机位点靶基因,发现增强子显著正调控靶基因的表达;保守性与GC含量分析表明增强子具有显著高的保守性与GC含量,并存在C-T/C-T/C-T-G模式的motif;增强子功能分析显示增强子显著富集于蛋白结合、酶结合、转录因子结合、RNA聚合酶Ⅱ结合等已知增强子功能,增强子GO与KEGG通路功能富集分析表明增强子靶基因显著参与细胞增殖、细胞凋亡、细胞周期调控和细胞迁移等肿瘤相关的生物进程与信号通路。结论:识别的肝细胞癌增强子具有显著高的保守性与GC含量,受多种转录因子调控,对其靶基因起正调控作用并且显著富集于肿瘤相关生物学进程与信号通路中。
增强子;肝细胞癌;Chip-seq;组蛋白修饰
肝细胞癌是死亡率最高的癌症之一,其发生往往与基因表达失调相关,因此解析参与肝细胞癌发生发展的调控关键因子及调控网络对于肝细胞癌研究具有重要意义[1]。增强子一般是几百碱基对长度的DNA片段,并能被多个转录因子占据,在基因调控中通过顺式调控原件对靶基因起到正调控作用。已有研究表明肝细胞癌中的增强子突变会导致增强子失活,进而影响靶基因的表达[2],因此探寻增强子在肝细胞癌中的调控网络,对于从转录水平了解肝细胞癌发生发展机制具有重要作用。增强子依据其活性分为活性增强子与失活增强子,而往往活性增强子是结合转录因子并行使下游调控功能的主要增强子类型。活性增强子一般处于能够被DNaseⅠ所切割的开放染色质区域DHS(DNaseⅠhypersensitive sites)中,开放染色质结构对于转录因子结合是必需的,所以DHS也被认为是一种识别增强子区域传统和有效的方法[3]。增强子相关的核小体的组蛋白末端共价修饰对于转录因子的招募具有重要作用[4],以往研究表明活性增强子存在显著的组蛋白修饰H3K27ac信号富集及高的H3K4me1/H3K4me3信号比值,并且H3K27ac修饰可以作为区分活性增强子和非活性增强子的标志[5-6]。我们通过增强子特征识别了肝癌细胞HepG2中的增强子,并对增强子序列功能进行了分析。通过整合来自ENCODE[7]的HepG2细胞系的3种组蛋白修饰(H3K27ac、H3K4me1、H3K4me3)数据及DHS数据,我们共识别了2254条肝癌细胞的增强子及1432个增强子的靶基因,分析了这些增强子的保守性、GC含量及结合转录因子富集情况,并对增强子及增强子的靶基因进行了GO与KEGG通路功能富集分析。结果发现HepG2中的增强子具有显著高的保守性与GC含量,并有大量转录因子富集,增强子功能分析显示增强子显著与蛋白结合、转录因子结合等增强子功能相关,而增强子靶基因GO与KEGG通路富集分析表明增强子靶基因显著参与细胞增殖、细胞凋亡、细胞周期调控、细胞迁移等肿瘤相关生物进程与信号通路。这些研究有望为进一步探讨肝细胞癌的基因调控及功能分析提供理论依据。
1 材料与方法
1.1 材料
组蛋白修饰和DHS的Chip-seq[8]数据来自ENCODE,包括peak文件和signal文件:H3K4me1(ENCFF635NAK、ENCFF159AXA),H3K4me3(ENCFF822KCS),H3K27ac(ENCFF558QIW、ENCFF542IPN) , DHS (ENCFF673GQN、ENCFF776TKB);基因组序列保守性数据来自UC⁃SC cons46way track;基因组GC含量数据来自UC⁃SC GC Percent track;转录因子结合位点数据来自 ENCODE Txn Factor(V3);肝脏基因表达谱的RNA-seq数据(RPKM)自GTEx[9]获得。
1.2 方法
1.2.1 HepG2中增强子的识别 首先用bwtool[10]和deeptools[11]对3种组蛋白修饰及DHS信号进行归 一 化 ,筛 选 2kb内 有 DHS、H3K27ac和H3K4me1 peaks,且H3K4me1的平均归一化信号高于H3K4me3的区域作为候选增强子区域,与参考基因组(GENCODE.V19)的蛋白编码基因比较,去除与蛋白编码基因区域及其蛋白编码基因转录起始位点上游1kb区域内有交集的增强子后,剩余的增强子作为最终识别的增强子。
1.2.2 保守性分析及GC含量 通过bwtool获得每个增强子的PhastCons分值,并计算每个增强子中心上下游100bp的DHS信号均值来表示该增强子的活性,将各个组织活性最高的前1000条增强子序列输入MEME,获得每个组织增强子的motif,参数如下:-revcomp-nmotifs 3-minw 10-evt 0.01-mod oops-dna。每个增强子中心及附近位点的GC含量通过bwtool获得。
1.2.3 增强子上转录因子结合位点识别 结合在增强子中心2kb以内的转录因子结合位点被认为对增强子有调控作用,每个增强子周围的转录因子结合位点数目被定义为该增强子上的转录因子富集程度。
1.2.4 增强子GO分析和靶基因定义及GO和KEGG信号通路分析 距离增强子100kb内的邻近蛋白编码基因被定义为该增强子的靶基因,并根据从GTEx获得的基因表达量数据得到每个增强子的靶基因表达量。用GREAT[12]预测每个组织增强子的功能(P<0.01),用 DAVID[13]对增强子靶基因进行GO和KEGG信号通路分析,以P<0.05为显著性阈值。
2 结果与讨论
2.1 增强子的识别
根据3种组蛋白修饰特征及DHS,我们共识别了2254条增强子。正如预期,增强子整体呈现高的H3K27ac信号,以及高的H3K4me1/H3K4me3信号比,且3种组蛋白修饰信号都呈双峰模式,即文献报道的peak-valley-peak模式[14]。组蛋白修饰信号在开放染色体区域附近显著增高,且DHS信号峰值位于增强子中心,这些结果说明我们找到的增强子符合活性增强子特征(图1)。
2.2 增强子保守性及GC含量分析
增强子做为一种顺式调控原件,在基因表达调控上起重要作用,因此,增强子序列往往较为保守。我们采用PhastCons计算了增强子的保守性分值,通过与随机位点相比较,发现增强子中心附近500bp内呈现显著高的保守性分值(图2A),且保守性相对增强子中心呈对称分布。相比于增强子来说,基因的保守性则在转录始位点(transcription start sites,TSS)下游呈现显著高的保守性,这与先前的研究结果一致。另外,为了确定这些增强子所具有的保守功能模块,我们用MEME工具对增强子进行了模体(motif)分析,结果显示其具有典型的C-T/C-T/C-T-G模式的motif(图2B)。先前研究表明,活性增强子普遍存在低甲基化现象[15],而甲基化与GC含量存在显著的相关性[16],为此,我们分析了增强子的GC含量(图2C)。正如预期,相比于人类基因组41.6%的背景GC含量,我们得到的HepG2增强子的GC含量(50.4%)显著高于人基因组平均GC含量(图2C),且GC含量在增强子中心附近200bp内富集最为显著。与之相反,基因的GC含量在TSS上游区域较低而在TSS附近区域呈现显著高的GC含量。这一结果说明我们获取的增强子存在潜在的甲基化倾向,并且已有研究表明肝细胞癌中DNA甲基化受抑制会导致参与肿瘤的生理调控及能量代谢平衡的AMPK信号通路发生改变[17]。

图1 增强子上下游2000bp内的3种组蛋白修饰及DHS归一化平均信号
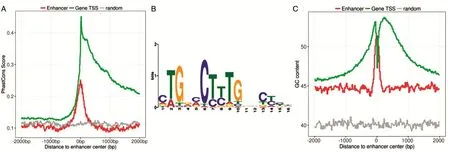
图2 增强子保守性、motif及GC含量分析结果
2.3 转录因子富集程度分析
活性增强子往往通过募集大量转录因子从而增强下游基因的表达,为此识别增强子上的转录因子结合情况对于解析增强子参与的转录调控具有重要意义。因此,我们将ENCODE收录的HepG2中的转录因子结合位点数据与本研究得到的增强子进行整合,发现有135个转录因子的9983个转录因子结合位点结合到了我们识别的增强子上,并且增强子的转录因子富集程度显著高于随机位点(P<2.2×10-16)(图3A)。其中,转录因子FOXA1在增强子中富集最明显,共有961转录因子结合位点。FOXA1能显著抑制癌细胞特异的基因表达[18],并有研究表明增强子上的FOXA1结合位点突变会导致增强子失活进而影响其靶基因的表达[2],这些暗示着肝细胞癌增强子上的FOXA1结合位点对于肝癌细胞的代谢及生物进程可能发挥着重要作用。

图3 增强子的转录因子富集程度与靶基因的表达量。
2.4 HepG2活性增强子靶基因识别及表达量分析
增强子作为一种顺式调控原件,其最主要的功能是正调控邻近基因的表达。为了验证我们识别的增强子的这一功能,通过与邻近已经注释的蛋白编码基因位置信息比较,共获得1432个增强子的靶基因。为了验证这些靶基因是否由于增强子的存在引起其表达上调,与随机基因的表达进行了比较,发现识别的增强子靶基因表达量显著高于随机基因(P<0.05)(图3B),这暗示本研究识别的肝细胞癌增强子对其靶基因的表达起到了明显的增强作用。
2.5 增强子GO分析及靶基因的GO和KEGG信号通路分析
通过使用GREAT获得增强子作为顺势调控元件的功能,可以看出增强子的分子功能显著与增强子功能相一致,如蛋白结合(4.10E-16)、酶结合(2.48×10-15)、转录因子结合(8.82×10-8)和RNA聚合酶Ⅱ结合(1.70×10-8)。值得注意的是,RNA聚合酶Ⅱ在增强子上的结合意味着肝细胞癌增强子同样可以招募RNA聚合酶Ⅱ从而转录出转录本,即增强子 RNA(enhancer RNA,eRNA),这类RNA的功能目前尚不清楚,有研究报道eRNA与增强子靶基因调控相关[19]。另外,通过对增强子靶基因的GO分析,发现增强子调控的靶基因显著参与细胞增殖、细胞凋亡、细胞周期调控、细胞迁移等肿瘤相关的生物进程(表1)。并且,在增强子靶基因的KEGG分析中发现其靶基因显著参与肿瘤相关的信号通路,如MAPK信号通路(在肿瘤细胞增殖、凋亡中发挥调控作用)、TNF信号通路(调控肿瘤内皮细胞因子活性)和癌症中的通路等(表2)。
2.6 结论
本研究整合了3种组蛋白修饰(H3K27ac、H3K4me1和H3K4me3)数据及DNaseⅠ高敏位点DHS信号,预测了2254个增强子,并获得1432个增强子的靶基因,结合转录因子结合位点数据,获得可能调控增强子的135个转录因子及9983个转录因子结合位点。在对增强子的保守性分析中获得了HepG2中的增强子可能发挥功能元件作用的C-T/C-T/C-T-G模式的motif。通过靶基因的表达量分析,表明预测得到的增强子能显著正调控基因表达。增强子的高GC含量意味着增强子存在潜在的甲基化调控模式,这与肿瘤的发生发展高度相关。对增强子的功能分析表明增强子发挥增强子已知的普遍功能,如蛋白结合、转录因子结合、RNA聚合酶Ⅱ结合等,其靶基因显著参与肿瘤相关的生物进程及信号通路。这些结果将为进一步研究肝细胞癌的基因调控及功能提供理论依据。

表1 增强子靶基因参与的肿瘤相关生物进程

表2 增强子靶基因参与的肿瘤相关KEGG信号通路
[1] Costentin C.Hepatocellular carcinoma surveillance[M].Paris:La Presse Médicale,2017.
[2] Huang D,Ovcharenko I.Identifying causal regulatory SNPs in ChIP-seq enhancers[J].Nucleic Acids Res,2015,43(1):225-236.
[3] Wang Y M,Zhou P,Wang L Y,et al.Correlation be⁃tween DNaseⅠhypersensitive site distribution and gene expression in HeLa S3 cells[J].PLoS One,2012,7(8):e42414.
[4] Heinz S,Romanoski C E,Benner C,et al.The selec⁃tion and function of cell type-specific enhancers[J].Nat Rev Mol Cell Biol,2015,16(3):144-154.
[5] Creyghton M P,Cheng A W,Welstead G G,et al.Histone H3K27ac separates active from poised enhanc⁃ers and predicts developmentalstate[J].Proc Natl Acad Sci USA,2010,107(50):21931-21936.
[6] Guenther M G,Levine S S,Boyer L A,et al.A chro⁃matin landmark and transcription initiation atmost promoters in human cells[J].Cell,2007,130(1):77-88.
[7] Raney B J,Cline M S,Rosenbloom K R,et al.EN⁃CODE whole-genome data in the UCSC genome brows⁃er(2011 update)[J].Nucleic Acids Res,2011,39(Data⁃base issue):D871-875.
[8] Jothi R,Cuddapah S,Barski A,et al.Genome-wide identification ofin vivo protein-DNA binding sites from ChIP-Seq data[J].Nucleic Acids Res,2008,36(16):5221-5231.
[9] Consortium G T. The genotype-tissue expression(GTEx)project[J].Nat Genet,2013,45(6):580-585.
[10]Pohl A,Beato M.bwtool:a tool for bigWig files[J].Bioinformatics,2014,30(11):1618-1619.
[11]Ramirez F,Dundar F,Diehl S,et al.deepTools:a flexible platform for exploring deep-sequencing data[J].Nucleic AcidsRes,2014,42(Web Serverissue):W187-191.
[12]McLean C Y,Bristor D,Hiller M,et al.GREAT im⁃proves functional interpretation of cis-regulatory regions[J].Nat Biotechnol,2010,28(5):495-501.
[13]Jiao X,Sherman B T,Huang da W,et al.DAVIDWS:a stateful web service to facilitate gene/protein list analysis[J].Bioinformatics,2012,28(13):1805-1806.
[14]Pundhir S,Bagger F O,Lauridsen F B,et al.Peakvalley-peak pattern of histone modifications delineates active regulatory elementsand theirdirectionality[J].Nucleic Acids Res,2016,44(9):4037-4051.
[15]Stadler M B,Murr R,Burger L,et al.DNA-binding factors shape the mouse methylome at distal regulato⁃ry regions[J].Nature,2011,480(7378):490-495.
[16]EhrlichM,Gama-SosaM A,HuangL H,etal.Amount and distribution of 5-methylcytosine in hu⁃man DNA from different types of tissues of cells[J].Nucleic Acids Res,1982,10(8):2709-2721.
[17]Sun Q,Xie Y,Wang G,et al.Identification of genes in HepG2 cells that respond to DNA methylation and histone deacetylation inhibitor treatment[J].Exp Ther Med,2014,8(3):813-817.
[18]Zhang G,Zhao Y,Liu Y,et al.FOXA1 defines can⁃cer cell specificity[J].Sci Adv,2016,2(3):e1501473.
[19]Melamed P,Yosefzon Y,Rudnizky S,et al.Transcrip⁃tional enhancers:Transcription,function and flexibility[J].Transcription,2016,7(1):26-31.
Identification and Bioinformatics Analysis of Enhancers in Hepatocellular Carcinoma HepG2
DING Ruo-Fan1,LI Yu-Peng1,ZHANG Yi-Ming1,ZHU Xiao-Dong1,HU Hai-Bi1,LIU Wen-Rong1,LI Ling2,GUO Zhi-Yun2*
1.School of Life Science and Engineering,Southwest Jiaotong University,Chengdu 610031;2.Department of Pathology,Third People's Hospital,Chengdu 610031;China
*Corresponding author,E-mail:zhiyunguo@gmail.com
Objective:To resolve the enhancer regulation network of hepatoma cell,the enhancers were identi⁃fied by integrating the features of the enhancers in the hepatoma cell HepG2,and GC content,regulation of tran⁃scription factors,identification of target genes and functional enrichment were analyzed.Methods:Enhancers in HepG2 were predicted by integrating Chip-seq data of histone modifications H3K27ac,H3K4me1 and H3K4me3 and of DNaseⅠ hyper-sensitivity sites.The average PhastCons score and GC content of each enhancer were calcu⁃lated to assess the conservation and GC content of the overall enhancers.ENCODE transcription factor bindingsites data were integrated to search for transcription factor-enhancer regulation.The enrichment analysis of GO and KEGG pathway was performed by using GREAT and DAVID on enhancers and the target genes of enhancers respectively.Results:A total of 2254 enhancers in HepG2 were predicted,and 1432 target genes of enhancers,135 transcription factors and 9983 transcription factor binding sites of enhancers were obtained.The enhancers in HepG2 significantly promoted the expression of target genes by comparing with random regions.The analysis of conservation and GC content showed that the enhancers were significantly conserved and had a remarkably high GC content,and the motif of enhancer was C-T/C-T/C-T-G.The analysis of the function enrichment of GO and KEGG pathway of enhancers showed that the target genes of enhancers were involved in cell proliferation,cell apoptosis,regulation of cell cycle and cell migration and other tumor related biological processes and signaling pathways.Conclusion:Enhancers in HepG2 were significantly conserved and had a remarkable high GC content enrichment,and they were regulated by a variety of transcription factors and played a positive role in regulation on their target genes,and were significantly enriched in tumor-related biological processes and signaling pathways.
enhancer;hepatocellular carcinoma;Chip-seq;histone modification
Q751;Q811.4
A
1009-0002(2017)04-0455-05
2017-02-10
中央高校基本科研业务费专项(2682016YXZT04);国家大学生创新性实验计划(201610613066);四川省大学生创新创业训练计划(2016095)
丁若凡(1991- ),男,硕士研究生,(E-mail)dingruofan1@foxmail.com
郭志云,(E-mail)zhiyunguo@gmail.com
10.3969/j.issn.1009-0002.2017.04.010
