稀土三元/TMAF体系催化二氧化碳与环氧丙烷交替共聚及机理研究
2017-01-09孟庆洋程瑞华侯侨丽刘柏平李佳佳
孟庆洋, 程瑞华, 侯侨丽, 潘 星, 刘柏平, 李佳佳
(华东理工大学化学工程联合国家重点实验室, 上海 200237)
稀土三元/TMAF体系催化二氧化碳与环氧丙烷交替共聚及机理研究
孟庆洋, 程瑞华, 侯侨丽, 潘 星, 刘柏平, 李佳佳
(华东理工大学化学工程联合国家重点实验室, 上海 200237)
在稀土三元催化体系(三氯乙酸钇-二乙基锌-甘油)中加入相转移剂四甲基氟化铵(TMAF),并用于催化二氧化碳和环氧丙烷交替共聚。采用核磁共振(1H-NMR)、凝胶渗透色谱(GPC)、差示扫描量热(DSC)、热重分析(TG)和原位傅里叶转换红外光谱(insituFT-IR)等对聚合过程和产物结构性能进行表征分析。采用基于密度泛函理论(DFT)的分子模拟方法对聚合起始步骤链增长机理进行探究。研究结果表明:稀土三元/TMAF催化体系可以在不改变聚合特征和共聚物结构性能的情况下,得到高达4 740.6 g/mol(1 mol Zn催化得到聚合物的质量)的催化活性,且将催化剂诱导期从100 min缩短至20 min。机理研究表明:环氧丙烷的插入步骤为反应的速控步骤,且该步骤所需克服的吉布斯自由能能垒随模型催化剂活性锌中心的自然成键轨道电荷的增加而降低,环状链增长机理中二氧化碳的插入并“返咬”成环所需克服的吉布斯自由能能垒很高,进一步证实了二氧化碳和环氧丙烷共聚反应更倾向于遵循链状链增长聚合机理形成聚碳酸亚丙酯。
二氧化碳; 环氧丙烷; 相转移剂; 稀土三元催化剂; 交替共聚机理
随着二氧化碳排放量的增加,温室效应日益加重,如何有效地回收及利用温室气体CO2变得尤为重要[1]。CO2作为一种理想的C1资源,具有廉价、无毒和不易燃等特点[2]。自从1969年Inoue等[3]首次发现CO2与环氧化合物在二乙基锌-水催化体系下可以合成可降解的脂肪族聚碳酸酯以来,不同催化体系的开发及改性成为该领域的研究热点[1,4-6]。经过科学家们几十年的努力,许多催化体系被开发并应用于催化CO2和环氧化合物聚合,最具代表性的催化体系包括双金属氰化物催化剂、金属Salen催化剂、金属羧酸盐催化剂和β-二亚胺催化剂等。
作为非均相催化剂的代表,稀土三元催化剂具有合成方法简单、聚合产物分子量高和制备价格低廉等优点。我国稀土资源丰富、品种齐全,为开发利用稀土类催化剂提供了便利的资源条件[7]。1991年沈之荃等[8]首次采用稀土三元催化剂实现了CO2和环氧丙烷(PO)的聚合反应,所得聚合产物链段中碳酸酯链节含量很高,且聚合产物具有分子量较高和分子量分布较窄等特点。Tan等[9]用三氟乙酸钇和二乙基锌分别替代了Y(P204)3和烷基铝合成了一种新型稀土三元催化剂,其催化效率进一步提高,且所得聚合产物链段中碳酸酯链节含量大于95%。随后,王献红等[10-11]详细研究了稀土三元催化剂催化CO2与环氧化物的共聚反应,用三氯乙酸钇代替三氟乙酸钇,进步提高了催化效率。王献红[12]还将稀土三元催化剂负载在硅胶或三氧化二铝等无机载体上,这种方法可以显著地提高催化剂活性,其反应机理研究表明[13]:在加入稀土盐后,稀土三元催化剂的金属锌中心电子云密度降低。2012年,王献红等[14]使用稀土三元催化体系与双金属氰化物催化体系进行复合并催化CO2与PO聚合,得到了聚碳酸酯-聚醚的无规聚合物。2005年侯召民等[15]使用环戊二烯基稀土催化剂成功催化了CO2与环氧环己烷的聚合,单位时间转换率(TOF)为1 000~2 000 h-1。随后,在环戊二烯基稀土催化剂与芳香族氢氧化物共催化CO2与环氧环己烷聚合研究中,虽然催化剂活性低(TOF为3.7~9.4 h-1),但聚合物链段中碳酸酯链节含量高达99%[16]。以上研究表明,稀土三元催化体系催化CO2和环氧化合物的研究引起了科学家的广泛关注,但如何在得到高催化活性的同时保持聚合物高碳酸酯链节含量是该领域的一大难点。
近年来,随着相转移催化在有机合成领域的快速发展,季铵盐在高分子聚合领域的应用潜力已得到科学家的认可。Liaw等[11]以四丁基溴化铵为相转移剂,成功合成了收率高且分子量大的均聚碳酸酯。但目前将相转移剂引入稀土三元催化体系的研究尚鲜见报道。本文为了进一步提高稀土三元催化体系催化CO2和PO聚合的催化活性并缩短催化剂诱导期,引入了具有更强吸电子效应的含氟相转移剂四甲基氟化铵(TMAF)。同时,采用基于密度泛函理论(DFT)的分子模拟方法,通过原位红外与DFT模拟相结合的方法对链状链增长和环状链增长机理进行了研究。研究结果表明:TMAF的加入可以使稀土三元催化剂更好地与PO配位并使其开环,降低了环氧化合物开环速控步骤对于聚合反应活性的影响。由于PO的插入为该聚合反应催化剂诱导期的决定因素,因此TMAF也可以通过加速PO配位缩短催化剂诱导期。
1 实验部分
1.1 主要原料
三氯乙酸钇(Y(CCl3COO)3)、甘油(glycerine)、二乙基锌(ZnEt2)、四甲基氟化铵(TMAF):分析纯,Sigma Aldrich公司;Davison955硅胶(SiO2):美国Grace公司;PO、甲苯、叔丁醇、丙酮:分析纯,国药集团化学试剂有限公司,其中PO在使用前经氢化钙回流8 h,并在氮气保护下进行保存。CO2:纯度为99.99%,伟创化学有限公司。
1.2 催化剂的制备
1.2.1 ZnEt2-glycerine二元催化剂的制备 首先将1 mL甘油在高纯氮气保护下加入到预先处理过的250 mL三口烧瓶中,然后将2 mL ZnEt2以0.5 mL/min的速率逐滴滴加到三口烧瓶中,反应温度保持0 ℃,在磁力搅拌下反应0.5 h,即可制得ZnEt2-glycerine二元催化剂,标记为C1。
1.2.2 ZnEt2-Y(CCl3COO)3-glycerine稀土三元催化剂的制备 首先将0.8 g Y(CCl3COO)3和PO加入到高纯氮气保护下预先处理过的250 mL三口烧瓶中,再将1 mL甘油加入混合溶液中。混合一段时间后,将2 mL ZnEt2以0.5 mL/min的速率逐滴滴加到三口烧瓶中,反应温度保持0 ℃,在磁力搅拌下反应0.5 h,即可制得ZnEt2-Y(CCl3COO)3-glycerine稀土三元催化剂,标记为C2。
1.2.3 ZnEt2-Y(CCl3COO)3-glycerine/TMAF稀土三元催化剂的制备 在上述制备的稀土三元催化剂中加入0.6 g TMAF,进一步搅拌0.5 h,制得ZnEt2-Y(CCl3COO)3-glycerine/TMAF稀土三元催化剂,标记为C3。
1.3 聚合反应
首先,将1 L高压釜在60 ℃下抽真空1 h,再将一定量的催化剂加入到高压釜并加入一定量的PO,然后通入CO2至压力为5.5 MPa,最后将反应温度设为70 ℃,转速设为350 r/min,反应8 h。聚合反应结束后,立即将反应釜冷却,并从气体进料口缓慢泄出剩余的CO2和PO混合气。待常压后向釜内加入乙醇水溶液终止反应,得到聚碳酸亚丙酯(PPC),50 ℃抽真空至恒重备用,其反应示意图如图1所示。以C1、C2和C3催化CO2和PO交替共聚得到的PPC分别标记为PPC1、PPC2和PPC3。
图1 CO2和PO的聚合反应Fig.1 Copolymerization of CO2 and PO
1.4 表征
1H-NMR光谱采用AVANCE 500核磁共振谱仪测定,溶剂为氘代氯仿,四甲基硅烷为内标,根据核磁共振结果,聚碳酸酯链节含量(Wpc)由公式(1)计算[17];数均分子量(Mn)和重均分子量(Mw)采用Waters-1515凝胶渗透色谱(GPC)测定,四氢呋喃为洗脱剂,聚苯乙烯为标样;玻璃化转变温度(Tg)采用Q200差示扫描量热仪测得,其升温速率为10 ℃/min,温度范围为10~80 ℃;热分解行为(TG5%:热失重95%时的温度)采用SDT Q600型热重分析仪测定,样品在氮气气氛下以10 cm3/min的升温速率从50 ℃加热到500 ℃;原位红外光谱采用Vertex70红外光谱仪测定,光谱范围为4 000~600 cm-1,分辨率 0.09 cm-1,测量速率为每分钟1张谱图。
(1)
1.5 计算方法
本研究模型体系中所有中间体的几何优化和过渡态搜索都基于Gaussian 09软件包进行,并采用Becke提出的梯度修正交换泛函与Lee、Yang以及Parr提出的梯度修正相关泛函相结合的非局域杂化泛函B3LYP,锌原子采用Lanl2TZ(Los Alamos National Laboratory second triple-Zeta)基组,碳、氢、氧原子采用6-311G(d,p)基组。
2 结果与讨论
2.1 聚合结果
不同催化体系对CO2和PO的交替共聚的影响如表1所示。从表1可以看出,TMAF的加入提高了聚合活性,但并没有改变聚合产物的性能,聚合物的碳酸酯链节含量大于 98%,重均分子量最高达到5.90× 105,Tg与热稳定性也维持与稀土三元催化体系聚合产物相同的特点。

表1 不同催化体系CO2和PO交替共聚的影响Table 1 Alterheating copolymerization of CO2 and PO over different catalysts
2.2 共聚产物结构表征
图2分别为PPC2和PPC3的原位红外谱图。PPC中C=O的主要红外吸收峰出现在1 750 cm-1处。由图2可以看出,在不加入TMAF时,稀土三元催化剂的诱导期较长,在100 min内PPC的生成量很少。但在加入一定量的TMAF后,诱导期大幅度缩短(诱导期约为20 min)。由于在CO2与PO交替共聚反应中,PO既作为反应单体也作为反应溶剂,TMAF的加入使稀土三元催化体系在PO中的溶解度大大提高,促进了PO与催化剂活性中心Zn-OR键的配位作用[18-19]。
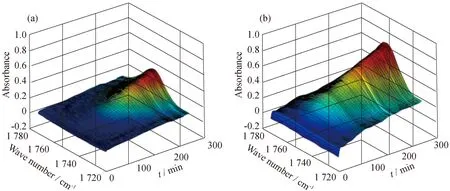
图2 PPC2(a)和PPC3(b)的动力学原位红外谱图Fig.2 In situ FT-IR of PPC2(a) and PPC3(b)
PPC的1H-NMR谱图如图3所示。从PPC3的1H-NMR谱图可以看出:1.31(d,3H;-CH3),4.17(m,2H;-H2C—),4.97(m,1H;-CH-)处均来自于PO的贡献。1.16和3.58 处分别为聚合物醚链节中的-CH3和-CH2CH-的化学位移。通过对比3种PPC可以看出:其结构非常相似,聚合物链段中的碳酸酯链节含量较高(>98%)。相比之下,在二元催化体系下得到的聚合产物中,1.16和3.58处出现了较明显的醚链节特征峰,而稀土三元催化体系中并没有出现,说明稀土配合物中Y(CCl3COO)3是提高聚合产物链段中碳酸酯链节含量的重要因素。
由PPC的GPC谱图(图4)可以看出,3种催化体系催化CO2和PO聚合得到的PPC全部具有较高的分子量和较窄的分子量分布。
2.3 链状与环状聚合机理研究
本文通过DFT模拟的方法进一步探究了该催化体系对聚碳酸酯链节具有较高选择性的原因。首先对反应单体CO2和不同构象的PO结构做了优化,结果如图5所示。
根据文献[19-21]中稀土三元催化剂催化CO2与PO共聚反应的研究,催化剂的活性中心可能为配位活化后的锌氧键。通过之前的实验研究[19]观察到,当二乙基锌和甘油的物质的量之比为2∶1时,催化体系的活性最高,推断二乙基锌和甘油催化体系的活性中心的结构如图6所示。
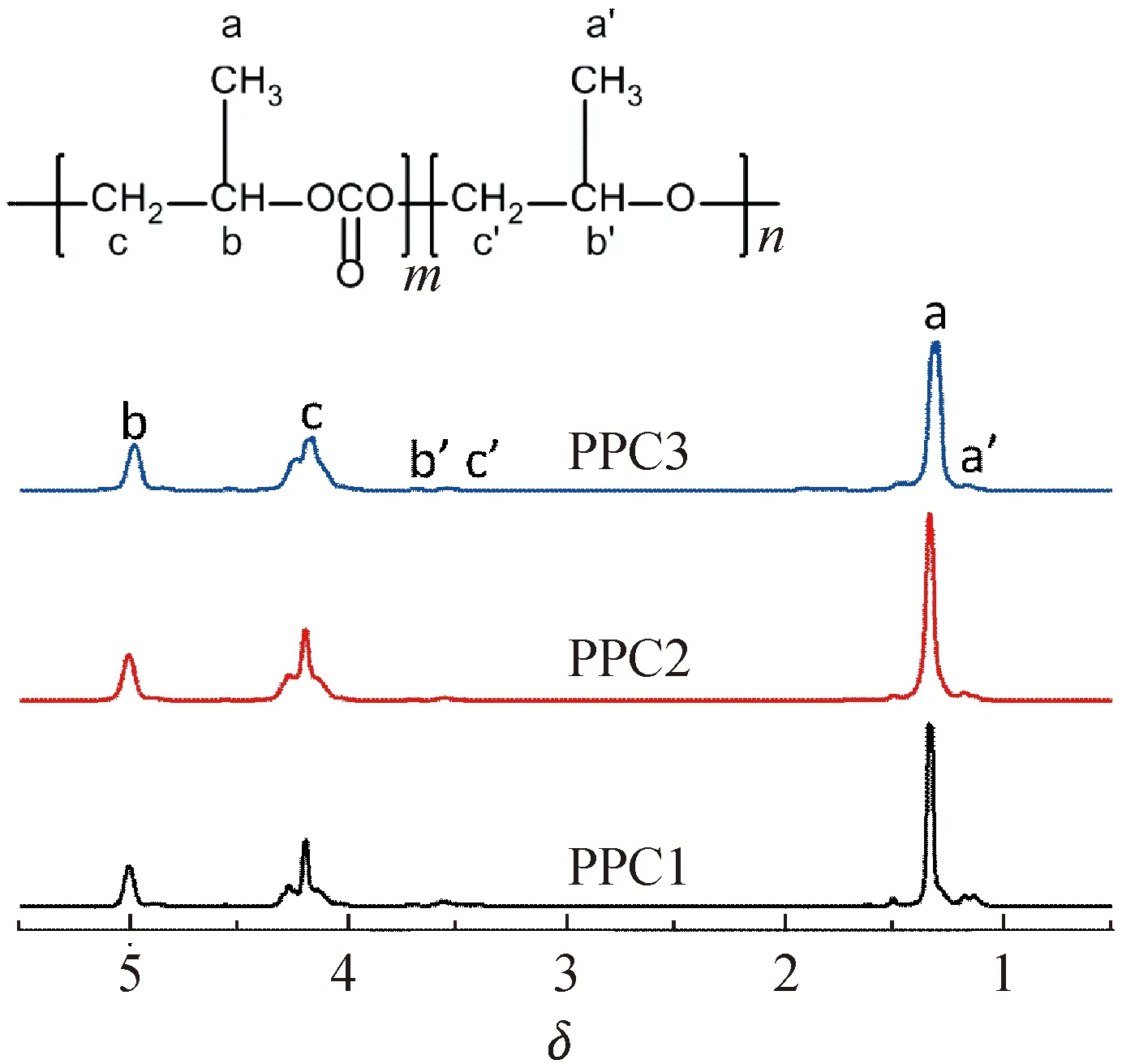
图3 PPC的1H-NMR谱图Fig.3 1H-NMR spectra of PPC
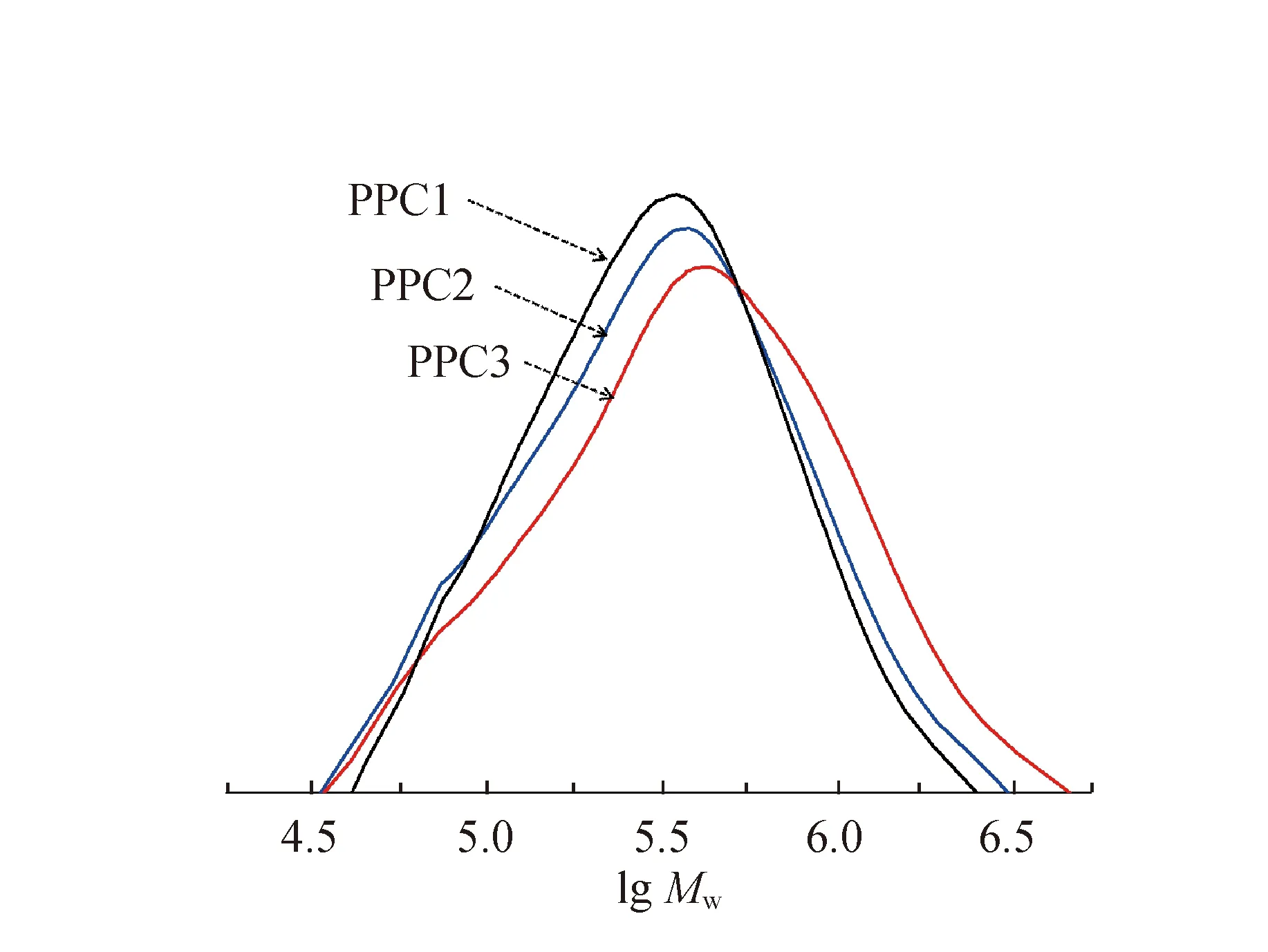
图4 PPC的GPC谱图Fig.4 GPC curves of PPC
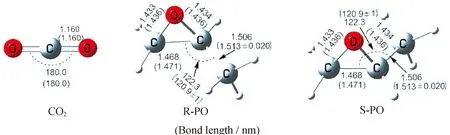
图5 CO2、R-PO和S-PO的结构优化Fig.5 Optimized structure of CO2,R-PO and S-PO
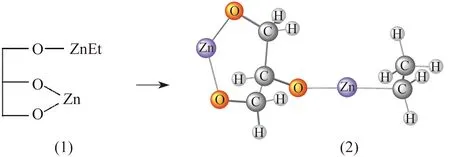
图6 简化的稀土三元催化体系结构模型(1)及其优化结构(2)Fig.6 Simplified structure of ternary rare earth metal complexes(1) and optimized structure(2)
首先设计了优化后的稀土三元催化体系催化CO2和PO共聚反应的反应路线,考查了不同位置的锌活性中心活化PO和CO2、不同的亲核物质活化PO的链状路径图及环状路径图,得到了反应机理的相关信息,如图7所示。链状链增长机理和环状链增长机理主要是由PO吸附金属锌中心的位置决定的。当PO吸附于直链上的锌中心时,PO和CO2的交替插入在支链上进行,遵循链状链增长机理;同理,当PO吸附于五元环上的锌中心时,链增长遵循环状链增长机理。
稀土三元催化体系催化CO2与PO交替共聚反应的中间结构计算结果如图7、图8所示。计算结果表明:所有的路径中,PO插入步骤的吉布斯自由能能垒是最高的,说明无论是链状链增长机理还是环状链增长机理,PO的插入均为速控步骤。对于二乙基锌体系,PO的插入是非常困难的,若能增强活性中心锌的缺电子性,使PO更容易开环,则可以大幅度提高聚合反应的活性。对比链状路径和环状路径,虽然环状路径中PO插的入吉布斯自由能能垒较低,但CO2插入的吉布斯自由能能垒较高,且随着插入原子的数目的增加,环张力将明显增大,CO2和PO交替插入形成环状路径的吉布斯自由能能垒也会随之增加。同时,由于环状链增长机理中,CO2插入发生分子内“反咬”所需克服的吉布斯自由能能垒较高,故此反应路径不可能发生。因此,稀土三元催化剂体系催化PO和CO2交替共聚将按照链状路径进行。对于链状路径,路径B1-H1(图7(a))及B1-H2(图7(b))中PO插入的吉布斯自由能能垒很高,相对而言路径B1-H3(图7(c))及路径B1-H4(图7(d))中PO插入的吉布斯自由能能垒低很多,因而反应很有可能按照路径B1-H3或路径B1-H4进行,具体情况还要根据CO2插入的吉布斯自由能能垒大小来决定。
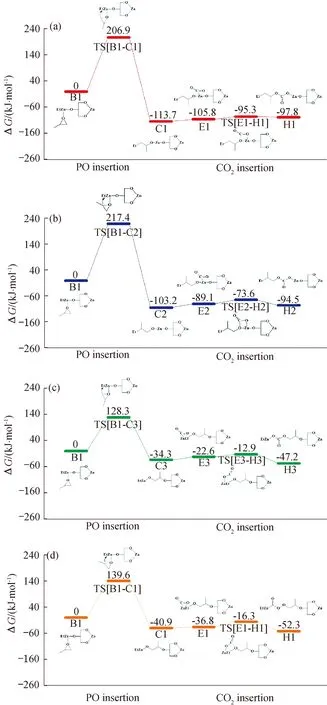
图7 不同配位的链状链增长路径计算结果Fig.7 DFT studies on the copolymerization of CO2 and PO to form linear chain growth structure
通过上述计算得到了稀土三元催化体系催化CO2与PO聚合初始的反应机理,如图9所示。PO上的氧原子首先与二乙基锌配体中的锌进行配位开环,通过自身异构化形成稳定的链状结构。随后CO2的C=O键与二乙基锌配体中的锌配位形成三元环,开环插入并完成链状链增长机理中PO与CO2的交替插入。对于环状链增长机理而言,随着CO2和PO的不断插入,环张力逐渐增大,反应所需克服的吉布斯自由能能垒不断升高,故环状链增长不可能发生。本文计算模拟得到的聚合机理与实验结果保持一致,PO和CO2继续插入的计算模拟有待进一步研究。
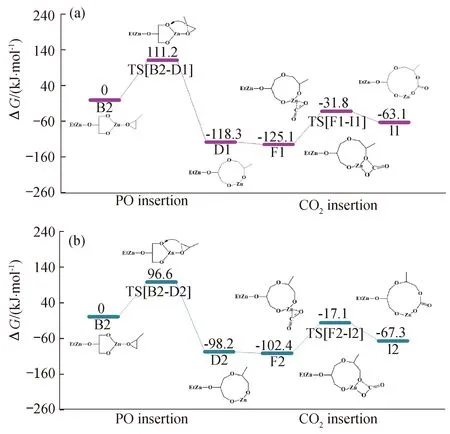
图8 不同配位的环状链增长路径计算结果Fig.8 DFT studies on the copolymerization of CO2 and PO to form cyclic chain growch structure
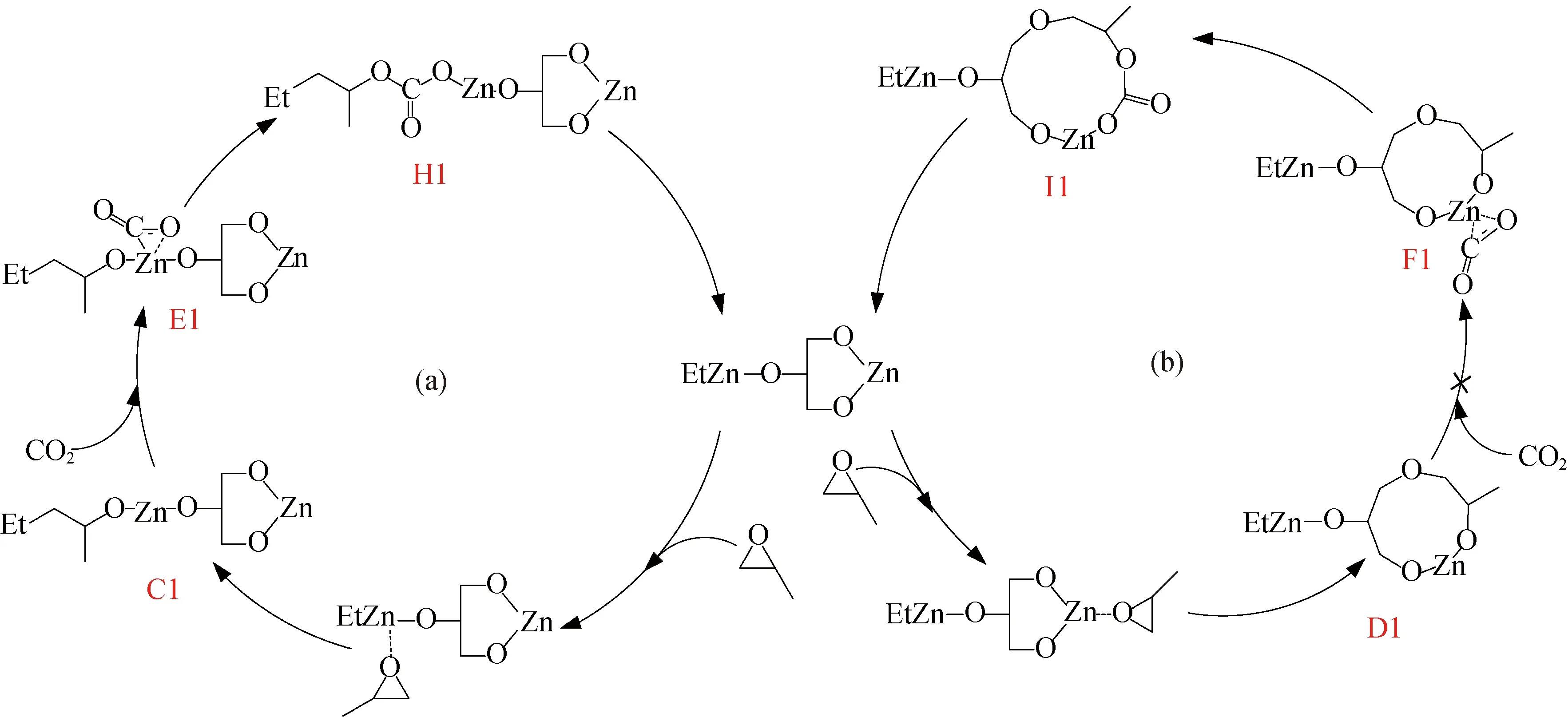
图9 CO2和PO交替共聚初始步骤的链状反应机理(a)及不可行的环状反应机理(b)Fig.9 Linear (a) and infeasible cyclic (b) chain growth mechanism of CO2 and PO copolymerization initial step
3 结 论
(1) 在稀土三元催化体系中引入相转移剂TMAF,可以将CO2和PO的聚合活性提高至4 740.6 g/mol,且将催化剂诱导期缩短近80%,但聚合产物仍保持与稀土三元催化体系聚合产物相同的结构和性能。
(2) 稀土三元体系催化CO2与PO交替共聚可以得到高选择性的链状聚碳酸酯(Wpc> 98%),PO的开环插入所需克服的吉布斯自由能能垒很高,为反应的速控步骤。
(3) 在环状链增长路径中,PO开环反咬所需要克服的吉布斯自由能能垒虽然相对较低,但CO2的进一步插入需要克服极高能量且不易进行。
[1] COATES G W,MOORE D R.Discrete metal-based catalysts for the copolymerization CO2and epoxides:Discovery,reactivity,optimization,and mechanism[J].Angewandte Chemie-international Edition,2004,43(48):6618-6639.
[2] WILLIAMS C K,HILLMYER M A.Polymers from renewable resources:A perspective for a special issue of polymer reviews[J].Polymer Reviews,2008,48(1):1-10.
[3] INOUE S,KOINUMA H,TSURUTA T.Copolymerization of carbon dioxide and epoxide with organometallic compounds[J].Die Makromolekulare Chemie,1969,130(1):210-220.
[4] GAO Z W,XIAO L F,CHEN J,etal.Recent advances in the synthesis of cyclic carbonates from carbon dioxide andepoxides[J].Chinese Journal of Catalysis,2008,29(9):831-838.
[5] MENG Q Y,PEPPER K,CHENG R H,etal.Effect of supercritical CO2on the copolymerization behavior of cyclohexene oxide/CO2and copolymer properties with DMC/Salen-Co(Ⅲ) catalyst system[J].Journal of Polymer Science Part A:Polymer Chemistry,2016.
[6] MENG Q Y,CHENG R H,Li J J,etal.Copolymerization of CO2and propylene oxide using ZnGA/DMC composite catalyst for high molecular weight poly(propylene carbonate)[J].Journal of CO2Utilization,2016(16):86-96.
[7] KISSLING S,ALTENBUCHNER P T,NIEMI T,etal.Zinc-catalyzed transformation of carbon dioxide[J].Zinc Catalysis:Applications in Organic Synthesis,2015:179-206.
[8] CHEN X,SHEN Z,ZHANG Y.New catalytic systems for the fixation of carbon dioxide:Copolymerization of carbon dioxide and propylene oxide with new rare-earth catalysts-RE(P204)3-Al(i-Bu) 3-R(OH)n[J].Macromolecules,1991,24(19):5305-5308.
[9] TAN C S,HSU T J.Alternating copolymerization of carbon dioxide and propylene oxide with a rare-earth-metal coordination catalyst[J].Macromolecules,1997,30(11):3147-3150.
[10] LIU B,ZHAO X J,WANG X H,etal.Copolymerization of carbon dioxide and propylene oxide with Ln(CCl3COO)3-based catalyst:The role of rare-earth compound in the catalytic system[J].Journal of Polymer Science Part A:Polymer Chemistry,2001,39(16):2751-2754.
[11] XIE D,WANG X H,ZHAO X J,etal.Influence of dial kylzinc in rare-earth ternary catalyst on the copolymerization of carbon dioxide and propylene oxide[J].Chinese Journal of Polymer Science,2005,23(6):671-674.
[12] QUAN Z,WANG X J,ZHAO X H,etal.Copolymerization of CO2and propylene oxide under rare earth ternary catalyst:Design of ligand in yttrium complex[J].Polyme,2003,44(19):5605-5610.
[13] MIN J D,ZHANG Y M,CHEN Y Y,etal.Catalytic mechanism of ternary rare-earth-metal coordination catalysts in the copolymerization of carbon dioxide and epoxide[J].Acta Polymerica Sinica,2009(3):233-237.
[14] DONG Y,WANG X H,ZHAO X,etal.Facile synthesis of poly(ether carbonate)s via copolymerization of CO2and propylene oxide under combinatorial catalyst of rare earth ternary complex and double metal cyanide complex[J].Journal of Polymer Science Part A:Polymer Chemistry,2012,50(2):362-370.
[15] CUI D,NISHIURA M,HOU Z.Alternating copolymerization of cyclohexene oxide and carbon dioxide catalyzed by organo rare earth metal complexes[J].Macromolecules,2005,38(10):4089-4095.
[16] CUI D,NISHIURA M,TARDIF O,etal.Rare-earth-metal mixed hydride/aryloxide complexes bearing mono(cyclopentadienyl) ligands:Synthesis,CO2fixation,and catalysis on copolymerization of CO2with cyclohexene oxide[J].Organometallics,2008,27(11):2428-2435.
[17] CHEN S,XIAO Z B,MA M Y.Copolymerization of carbon dioxide and epoxides with a novel effective Zn-Ni double-metal cyanide complex[J].Journal of Applied Polymer Science,2008,107(6):3871-3877.
[18] 曲荣君,孙昌梅,王春华,等.相转移催化在高分子化合物合成中的应用[J].催化学报,2003,24(9):716-724.
[19] PAN X,LIU Z,CHENG R,etal.Experimental and theoretical studies on CO2and propylene oxide(PO) copolymerization catalyzed by ZnEt2-glycerine-Y(CCl3COO)3ternary catalyst[J].Journal of Organometallic Chemistry,2014,753:63-71.
[20] PAN X,LIU Z,CHENG R,etal.Mechanism for alternating copolymerization of CO2and propylene oxide in diethylzinc-water catalytic system:A DFT study[J].Journal of CO2Utilization,2013(2):39-48.
[21] PAN X,LIU Z,CHENG R,etal.Insight into the reaction mechanisms between CO2and epoxides over Zn(Ⅱ) phenoxide catalytic system——A DFT study[J].Journal of Organometallic Chemistry,2015,775:67-75.
Alternating Copolymerization and Theoretical Study on CO2and Propylene Oxide over Ternary Rare Earth Metal/TMAF Complexes
MENG Qing-yang, CHENG Rui-hua, HOU Qiao-li, PAN Xing, LIU Bo-ping, LI Jia-jia
(State Key Laboratory of Chemical Engineering, East China University ofScience and Technology, Shanghai 200237, China)
The copolymerization behavior of CO2and propylene oxide(PO) over ternary rare earth metal complex(ZnEt2-Y(CCl3COO)3-glycerine/tetramethylammonium fluoride(TMAF)) were investigated systematically.The copolymerization process and chemical properties were characterized by1H-Nuclear Magnetic Resonance(1H-NMR),Gel Permeation Chromatography(GPC),Differential Scanning Calorimetry(DSC),Thermal Gravity(TG) and in-situ Fourier Transform Infrared Spectrometer(in situ FT-IR).The initial step of CO2and PO insertion was calculated by Density Functional Theory(DFT) method.Results showed that the application of TMAF in ZnEt2-Y(CCl3COO)3-glycerine catalyst dramatically increased the catalytic activity up to 4 740.6 g/mol(mass of polymer for 1 mol Zn) and decreased the catalytic induction period from 100 min to 20 min.The theoretical results demonstrated that the rate-determining step of CO2and PO copolymerization was the ring-opening/insertion period of PO.The Gibbs free energy of the rate-determining step decreased with the increase of natural bonding orbital of the zinc metal center.In addition,the Gibbs free energy of CO2insertion to form “back-bite” cyclic chain growth structure was high.Thus,the copolymerization of CO2and PO might follow the linear chain growth mechanism to obtain poly(propylene carbonate).
carbon dioxide; propylene oxide; phase transfer agent; ternary rare earth metal complexes; alternating copolymerization mechanism
1008-9357(2016)04-0424-008
10.14133/j.cnki.1008-9357.2016.04.008
2016-07-12
上海市浦江人才计划(16PJD016)
孟庆洋(1987-),男,山东威海人,博士,主要研究方向为二氧化碳与环氧化合物共聚合成聚碳酸酯的实验及机理研究。E-mail:13916726923@163.com
程瑞华,E-mail:rhcheng@ecust.edu.cn
O 623,O 662
