人参化学成分的药物代谢动力学研究△
2016-09-25杨秀伟
杨秀伟
(北京大学 天然药物及仿生药物国家重点实验室,北京大学 药学院 天然药物学系,北京 100191)
人参化学成分的药物代谢动力学研究△
杨秀伟*
(北京大学 天然药物及仿生药物国家重点实验室,北京大学 药学院 天然药物学系,北京100191)
人参广泛的治疗和保健作用已有许多研究,其主要有效成分人参皂苷调节多种生理活动。生物和环境因素可能影响人参皂苷的功效。人参皂苷的药代动力学和代谢研究结果证明:①人参皂苷在胃肠道的吸收性随分子结构中糖基化程度不同而不同;②在人源肠Caco-2细胞单层模型,多糖基化原形人参皂苷的膜渗透性差是限制其口服吸收不良的主要原因;③人参皂苷在被吸收进入循环系统之前,主要被代谢为相应的次皂苷和/或皂苷元;④某些人参皂苷在肠道内由肠内细菌所致生物转化产生的生物活性中间体和/或终末产物,能够反映它们的生物活化作用,提示人参皂苷很可能是前药;⑤原形人参皂苷及其脱糖基代谢产物从体内清除。本文概述了人参皂苷生物转化和/或代谢以及药代动力学的最新研究进展,为明确人参的药效物质基础提供了循证科学依据。
人参;人参皂苷;药物代谢动力学
传统中药人参PanaxginsengC.A.Meyer系驰名中外的大补要药,除“独参汤”、人参皂苷-Rg3(商品名如“参百益Ginsenbesty”)外,以根和根茎、茎叶、果实等为原料的中成药或保健品均有问世,用于各种目的的应用。由于人参含有多样性的化学成分[1-3],决定了其具有多样性的生物学活性和药理学作用[4],后者又与人参化学成分的药物代谢动力学(pharmacokinetics,简称为药动学;PK),即其体内过程,包括吸收(absorption)、分布(distribution)、代谢(metabolism)、排泄(excretion)、毒性(toxicity)(简称为ADMET)等密切相关,研究人参化学成分的ADMET性质成为现代科学“循证”人参价值的策略之一,其能够在分子水平上提供人参生物学活性和药理学作用的信息。伴随人参化学成分研究的不断深入及其动物、人体内人参化学分子及生物转化和/或代谢产物分析技术的不断进步,人参PK研究取得了许多进展,及时总结这些成果或信息,有利于人参研究的集约化健康发展。本文不但总结直接来源于人参的化学成分的PK,而且包括那些来源于三七Panaxnotoginseng(Burk.)F.H.Chen和西洋参PanaxquinquefoliumL.、且在人参中含有的化学物质的PK。
1 肠内生物转化
人参皂苷(G)-Rb1是人参皂苷中的主要成分之一,亦是20(S)-原人参二醇[20(S)-protopanaxadiol;20(S)-PPD]型皂苷的典型代表,因此,不但研究的比较早,而且研究的比较深入。给无菌大鼠(germfree rat)灌胃G-Rb1[5],在肠道和盲肠中检测不到人参皂苷化合物K(G-CK)。给药后7h,G-Rb1在肠道和粪便中的累积回收率为90%以上;给药后15h的累积回收率为70%以上;给小鼠和普通大鼠灌胃G-Rb1或人口服G-Rb1,血浆中检测不到G-Rb1。但是,如果给无菌大鼠在肠道内定植真杆菌(Eubacterium)sp.A-44获得悉生大鼠(gnotobiotic rat)再灌胃给予G-Rb1,给药后7h在肠道和累积粪便中可检测到大量的G-Rb1和G-CK;给药后15h仅检出相当量的转化产物G-CK和少量的转化产物G-Rd,而无原形G-Rb1。同时,在悉生大鼠血浆中可检测到G-CK。此证明肠内细菌具有转化G-Rb1的能力。将大鼠分为林可霉素(lincomycin)按4.8、0.12g·kg-1两个剂量处理组和无处理对照组[6],取处理组大鼠0、6、12、24、48h的粪便悬浮液与G-Rb1共温孵培养,在0.12g·kg-1剂量处理组检测到G-Rd和G-F2,但在4.8g·kg-1剂量处理组仅检测到G-Rd,提示林可霉素改变了大鼠肠内菌丛构成,对G-Rb1的转化程度不同。将G-Rb1与青年人的新鲜粪便共温孵培养[7],G-Rb1迅速被转化,至培养的8h内已完全转化,转化产物有G-Rd、G-CK和G-F2;至24h内,G-Rd消失,可检出20(S)-PPD;至48h后,仅能检出G-CK和20(S)-PPD,两者的摩尔比为1∶1,由此总结G-Rb1的转化过程为G-Rb1→G-Rd→G-F2→G-CK→20(S)-PPD。以1~60岁年龄段的58人的粪便为肠内细菌来源进行G-Rb1的转化研究,发现男性和女性的肠内细菌转化产生G-CK的能力基本相同,而以30~39岁的转化率最高。Shen等[8]分析了健康志愿受试者肠道转化G-Rb1的途径。将G-Rb1与肠内菌丛共温孵培养,转化产生G-Rd、F2、CK和绞股蓝皂苷(GY)XVII、LXXV等5个转化产物,G-Rb1→GY-XVII→GY-LXXV→G-CK转化途径迅速,为中间体过程,产物量少;G-Rb1经过GY-XVII→G-F2(主要产物)→GY-LXXV(微量产物)过程产生G-CK,G-Rd和GY途径存在年龄、性别上的个体差异。G-Rb1表现为在强酸条件下不稳定,在模拟胃液条件下迅速转化为G-F2、Rg3、Rh2和CK,但没有产生GY-XVII和LXXV,提示G-Rb1转化为GY-XVII和LXXV有赖于消化道细菌介导的酶促反应。文献[5-6]报道了β-D-葡萄糖苷酶在肠内菌转化G-Rb1上的作用。Bae等[9]报道真杆菌(Eubacteriumsp.)、链球菌(Streptococcussp.)和双歧杆菌(Bifidobacteriumsp.)水解龙胆二糖(gentiobiose)的能力比水解槐糖(sophorose)的能力强,经过G-Rd中间体转化G-Rb1为G-CK;梭杆菌(Fusobacterium)K-60水解槐糖的能力比水解龙胆二糖的能力强,经过GY-XVII中间体转化G-Rb1为G-CK。将G-Rb1与大鼠盲肠内容物共温孵培养[10],至培养的0.5h可检测到转化产物GY-XVII,G-Rd、F2和CK,主要转化产物为G-Rd,至培养6h的主要转化产物为G-CK;在大鼠大肠,亦能检测到这4个转化产物。Akao等筛选了29种人肠内细菌对G-Rb1的转化,其中仅有真杆菌sp.A-44有转化G-Rb1的能力,G-Rd和CK的产率分别为16.8%和70.3%;亦有报道口腔普菌(Prevotellaoris)是转化G-Rb1的肠内活性菌之一[11-12]。G-Rb1的肠内菌生物转化途径如图1。
将G-Rb2与大鼠盲肠内容物一起温孵培养,亦可检测到转化产物G-Rd、F2和CK等,至共温孵培养6h的主要转化产物为G-CK[10]。真杆菌、链球菌和双歧杆菌经过G-Rd中间体转化G-Rb2为G-CK[9]。G-Rb1与G-Rb2在大鼠肠内容物中亦可转化产生少量的C-25过氧化物[10]。
人肠内细菌可转化G-Rc为20-O-[α-L-呋喃阿拉伯糖基-(1→6)-β-D-吡喃葡萄糖基]-20(S)-原人参二醇和G-CK[13]。Bae等报道[14],人肠内细菌可转化G-Rc产生G-Rd、F2、CK、Mb、Mc和20(S)-PPD,所有受试人粪便菌丛皆能转化G-Rc为G-CK和20(S)-PPD,后者为主要转化产物。人粪便中的拟杆菌(Bacteroidessp.)、真杆菌和双歧杆菌转化能力较强,双歧杆菌K-103和真杆菌A-44经过G-Rd中间过程转化G-Rc为G-CK;拟杆菌HJ-15和双歧杆菌K-506经过G-Mb中间过程转化G-Rc为G-CK。
人肠内细菌口腔普菌可将G-Rd转化为3个产物,其中之一为G-CK[11-12]。从上述G-Rb1和Rb2的肠内菌转化产物中间体G-Rd亦可继续转化为相应的转化产物。
人肠内菌丛可将20(S)-G-Rg3转化为20(S)-G-Rh2[15-16]和20(S)-PPD[16],主要转化产物为20(S)-G-Rh2[16]。20(S)-G-Rg3迅速转化为20(S)-G-Rh2和20(S)-PPD,其转化速率是20(R)-G-Rg3转化为20(R)-G-Rh2和20(R)-PPD的19倍。从肠内菌丛中分离出的拟杆菌、真杆菌和双歧杆菌经过G-Rh2中间体转化G-Rg3为PPD,但梭杆菌仅能将G-Rg3转化为G-Rh2。生晒参和水参中不含G-Rg3,原人参二醇型皂苷转化为G-Rg3,在较大程度上依赖于环境条件。在60℃酸性条件下,原人参二醇型皂苷转化为G-Rg3和Δ20-G-Rg3;在中性条件下,G-Rb1、Rb2、Rc不能转化为这些产物;在酸性条件下的转化随着温孵时间和温度的增加而增加,最适转化温度为60℃、温孵5h。人肠内菌丛可转化G-Rg3和Δ20-G-Rg3为G-Rh2和Δ20-G-Rh2,其中的拟杆菌、双歧杆菌和梭杆菌转化G-Rg3为G-Rh2的能力较强[17]。
G-Rb1、Rb2、Rc、Rd、Rg3皆为原人参二醇型皂苷,其在肠内的转化程度依赖于酶的有无和活力[6]。因此,有研究者[18]对转化酶进行了筛选,以期获得高产率的转化产物。从米曲霉(Aspergillusoryzae)获得的粗乳糖酶(lactase)、β-半乳糖苷酶(β-galactosidase)、从绿色木霉(Trichodermaviride)获得的纤维素酶(cellulase)等能分别水解原人参二醇型皂苷高产G-F2、CK和Rd;从青霉(Penicilliumsp.)获得的乳糖酶能水解原人参二醇型皂苷产生主要产物G-Rd和G-Rg3、CK。
从已有的研究结果,判断肠内细菌所致上述原人参二醇型皂苷的转化是一个对原形化合物的“激活”过程,如对肿瘤细胞的细胞毒活性,G-CK>20(S)-G-Rh2≥20(S)-G-Rg3>G-Rb1>Rb2[15];对肿瘤L1210、P388、A549、Me180细胞株的细胞毒活性,20(S)-PPD>20(S)-G-Rh2>20(S)-G-Rg3>G-Rb1,在同样的条件下,20(R)-PPD、20(R)-G-Rh2、20(R)-G-Rg3和G-Rb1没有显示出细胞毒活性;对大鼠胃H+/K+ATPase活性,20(S)-G-Rh2和20(S)-G-Rg3都显示出抑制作用,而原形化合物G-Rb1没有显示出抑制作用[16];对于幽门螺杆菌(Helicobacterpylori)生长,无论是20(R)-PPD还是20(S)-PPD都显示出生长抑制活性[16-17]。G-Rc的转化产物G-CK、Mc和20(S)-PPD对肿瘤L1210和P388细胞株皆有明显的细胞毒活性,G-CK和Mc还有抗过敏和抗炎活性,原形化合物G-Rc没有显示活性[14]。G-Rh2亦显示出抗过敏活性[19]。
G-Re是人参皂苷中的主要成分之一,亦是20(S)-原人参三醇[20(S)-protopanaxatriol;20(S)-PPT]型皂苷的典型代表,不但研究的比较早,而且研究的比较深入。人肠内细菌可转化G-Re产生G-Rg1、Rg2、Rh1、F1和20(S)-PPT[20]。将G-Re与人肠内菌丛温孵培养3h,仍以原形状态存在;至培养6、9、12h,出现转化产物G-Rg1;至培养24h,G-Re和G-Rg1消失佚尽,仅能检出转化产物20(S)-PPT[13]。当将G-Re与从人肠内细菌丛中分离得到的一株菌株口腔普菌共温孵培养时,产生3个转化产物,推测其中的2个转化产物是G-Re的氧化转化产物。转化产物中未发现20(S)-PPT,但可检出G-Rg1[11]。此外,人肠内细菌丛中的双歧杆菌K-103转化G-Re为G-Rg1;双歧杆菌K-525和拟杆菌HJ15转化G-Re为G-Rg1和Rh1;拟杆菌JY6转化G-Re为G-Rg1、Rh1、F1和20(S)-PPT;梭杆菌K-60转化G-Re为G-Rg1、Rh1和F1[20]。双歧杆菌Int57和SJ32经过G-Rg2中间体将G-Re转化为G-Rh1;黑曲霉(Aspergillusniger)经过G-Rg1中间体将G-Re转化为G-Rh1;宇佐美曲霉(Aspergillususamii)转化G-Re为G-Rg2[21]。
人肠内细菌转化G-Rg1较难,转化反应发生的时间较长,可将其转化为G-Rh1、G-F1和20(S)-PPT。将G-Rg1与人肠内细菌温孵培养,至培养的24h以后原形化合物显著减少,至培养的48h完全消失,但24h后出现转化产物G-Rh1,至培养的48h达最大,以后缓慢下降。至培养的48h,20(S)-PPT开始出现并逐渐增加。大约有20%的G-Rg1转化为20(S)-PPT。另有研究证明,当将G-Rg1与人肠内细菌温孵培养3h,即有少量的G-Rh1和20(S)-PPT产生,绝大部分的G-Rg1没有被转化;温孵培养6h,已检测不出原形化合物和G-Rg1,有少量的G-F1,绝大部分是20(S)-PPT;而在12、24h,仅能检出20(S)-PPT[7,11,13,22]。肠内菌中的口腔普菌不能水解G-Rg1[12]。这些转化上的差异是由于不同来源的人肠内细菌组成不同造成的还是由于代谢条件不同造成的,有待深入研究,阐明其缘由是十分有意义的。如果是肠内细菌组成不同,以转化结果为先导,可能寻找到转化活性特异或转化活性更高的人肠内细菌。
将20(S)-G-Rg2与大鼠胃液温孵,可检出转化产物20(S)-G-Rh1、20(S)-PPT和24H-25-羟基-20(S)-G-Rg2。将它们再与大鼠肠液温孵培养,每个又可产生两个转化产物[23]。20(S)-G-Rh1在人肠内菌丛温孵培养体系中可转化为20(S)-PPT[20]。
绝大部分的人参皂苷的生物转化研究主要围绕单体化合物进行,Wang等[24]报道了红参提取物的人肠内菌丛转化,鉴定出30个转化产物,主要是脱糖基产物,并首次发现了人参皂苷的乳酸酯转化产物。
2 肠吸收、生物利用度
2.1人源肠Caco-2细胞单层及其他模型
人源肠Caco-2细胞单层模型,已用来评价药物的吸收特性。G-Ra3在Caco-2细胞单层模型双向转运的表观渗透系数(Papp)值在10-7~10-8数量级,预测其经肠道吸收不良[25]。
G-Rb1在Caco-2模型中,摄取和转运没有发生饱和现象,从顶端(AP)到底端(BL)的转运随浓度增加而呈线性增加,提示其过膜转运为被动扩散;转运的Papp值为(5.90±1.02)×10-8cm·s-1(C0=1 mg·mL-1),吸收过程不受细胞膜内P-糖蛋白(P-gp)和多药耐药相关蛋白(MRP)外排载体的调控,在Caco-2细胞的细胞摄取量为(0.77±0.03)μg·mg-1蛋白(C0=1 mg·mL-1),提示其经肠道吸收较差;胃液的酸性环境、大肠菌丛产生的酶及肝脏的首过作用皆对其口服吸收产生影响,但肠道黏膜的透过性低是其口服吸收差的主要影响因素[26-27]。总皂苷中的G-Rb1与单体G-Rb1给药相比,在Caco-2细胞模型中的吸收特性无明显差异,提示其他成分对G-Rb1的吸收特性无明显影响[27]。用糖尿病大鼠血清温孵Caco-2细胞单层,可促进细胞间通透性,导致G-Rb1吸收性增加[28]。
G-Rb2在Caco-2细胞单层模型中,双向转运的Papp值在10-6~10-7数量级,表明其经肠道吸收较差;Papp(BL→AP)/Papp(AP→BL)为9.63,表明外排大于吸收,提示可能有外排转运蛋白参与G-Rb2的转运过程[29]。
G-Rb3在Caco-2细胞单层模型中,双向转运的Papp值在10-6数量级,表明其在肠道具有中等程度的吸收;Papp(BL→AP)/Papp(AP→BL)为1.86,表明外排大于吸收,提示可能有外排转运蛋白参与G-Rb3的转运过程[30]。受这两个因素影响,G-Rb3可能表现为吸收性不良。
G-Rc和G-Rd在Caco-2细胞单层模型双向转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良[25]。
G-Rg3在Caco-2细胞单层模型双向转运的Papp值在10-7~10-8数量级,预测其经肠道吸收不良[25]。G-Rg3的摄取是时间及浓度依赖性的,其被动转运系数Kd为0.07 nmol·h-1·mg-1·port-1,最大摄取速率Vmax为3.32 nmol·h-1·mg-1·port-1,Km为16.31 μmol·L-1;在加入代谢抑制剂叠氮化钠及2,4-二硝基苯酚时摄取速率明显降低[31]。提示其摄取转运可能消耗能量。
G-Rh2在Caco-2细胞单层模型双向转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良[25];其吸收具有时间、pH值和浓度依赖性,在4 ℃时符合一级速率方程,在37 ℃、pH 7.4时符合二级速率方程,代谢抑制剂和低温会使其吸收速率降低,当细胞外pH 7.0时G-Rh2达到最大吸收,动力学分析表明它可能是P-gp的1个底物[32]。Gu等[33]探讨了G-Rh2吸收不良的原因。Caco-2细胞单层模型中,在4 ℃,G-Rh2在1~50 μmol·L-1的吸收转运是线性的,但在37 ℃、浓度超过10 μmol·L-1出现饱和现象;在37 ℃、浓度为10 μmol·L-1时,前60 min的吸收转运是线性的;240 min,20(S)-G-Rh2和20(R)-G-Rh2分别为2 173.70和979.38 ng·min·μg-1;转运的Papp值在10-7~10-8数量级,外流率在1.5以上。在肠灌流模型,至灌注的3 h,20(S)-G-Rh2和20(R)-G-Rh2的吸收分数分别为15.7%和9.1%。载体转运是G-Rh2的主要吸收机制,它是ABC转运体(ATP Binding cassettee transporters)的基质。ABC转运体介导的外流和难过膜性,是G-Rh2吸收不良的主要原因。在立体异构体上,20(R)-G-Rh2的吸收性低于20(S)-G-Rh2差向异构体[33]。顾轶等[34]研究了20(R)-G-Rh2的大鼠肠吸收动力学,给药后的十二指肠、空肠、回肠和结肠肠袢的曲线下面积(AUC)0→6h分别为(207±88)、(301±49)、(157±93)、(172±68)ng·mL-1·h-1,吸收速率常数Ka分别为(3.9±0.4)、(1.6±0.4)、(1.9±0.8)、(1.1±0.9)·h-1,达峰时间tmax分别为(0.44±0.13)、(1.75±0.50)、(1.13±0.63)和(2.00±1.41)h,达峰浓度分别为(82±17)、(110±11)、(36±8)和(43±7)ng·mL-1,说明20(R)-G-Rh2肠道吸收困难,十二指肠和空肠是它吸收的主要部位。
G-F2在Caco-2细胞单层模型,从AP→BL转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良;从BL→AP转运的Papp值在10-5~10-6数量级[25]。
以上是原人参二醇型皂苷的吸收研究,某些该类型皂苷在肠内转化为G-CK和PPD。G-CK和PPD在Caco-2细胞单层模型双向转运的Papp值在10-5~10-6数量级,预测其经肠道吸收良好[25],G-CK的吸收转运呈线性浓度依赖性,其机制为被动扩散[35]。值得注意的是如G-Rb1、Rb2本身对P-gp无抑制作用,但其肠内菌转化产物G-CK[36-37]和PPD[36]是P-gp的抑制剂,与P-gp基质药物联合口服用药可能存在药物相互作用的风险,需慎重。另一方面,与P-gp抑制剂合用,能增加人参皂苷的吸收。如在L-MDR1细胞模型,G-Rb1、Rb2、Rc、Rd、Rg3的外流率大于3.5,与P-gp抑制剂五味子木脂素合用,能明显降低它们的外流率和提高它们的摄取率[38]。
G-Re是人参中原人参三醇型皂苷的代表性成分之一,在Caco-2细胞单层模型双向转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良[25]。
G-Rf和20-葡萄糖基-G-Rf在Caco-2细胞单层模型双向转运的Papp值在10-7~10-8数量级,预测其经肠道吸收不良,但G-Rf要好于20-葡萄糖基-G-Rf[25]。
G-Rg1、G-Rg2和三七皂苷R1(NG-R1)在Caco-2细胞单层模型双向转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良[25]。另有报道[27],G-Rg1在胃液酸性环境下易被破坏,在近中性环境内基本保持稳定,大肠内容物中易降解,在小肠内容物中相对稳定。在Caco-2细胞单层的摄取受温度的影响,但pH的变化及环孢菌素A的加入对其摄取均无显著性影响;摄取和转运没有发生饱和现象,从AP到BL的转运随浓度增加而呈线性增加,提示其过膜转运为被动扩散;转运的Papp值为(2.59±0.17)×10-7cm·s-1(C0=1 mg·mL-1),吸收过程不受细胞膜内P-gp和MRP外排载体的调控,在Caco-2细胞的细胞摄取量为(1.07±0.16)μg·mg-1蛋白(C0=1 mg·mL-1),提示其经肠道吸收较差;胃液的酸性环境、大肠菌丛产生的酶及肝脏的首过作用皆对其口服吸收产生影响,但肠道黏膜的透过性低是其口服吸收差的主要影响因素。总皂苷中的G-Rg1与单体G-Rg1给药相比,在Caco-2细胞模型中的吸收特性无明显差异,提示总皂苷中的其他成分对G-Rg1的吸收特性无明显影响。李昊等[39]应用不同肠段的肠管外翻囊模型亦研究了P-gp抑制剂对G-Rg1的吸收影响,发现在回肠和空肠中G-Rg1的吸收良好,且无明显差异,P-gp抑制剂维拉帕米对G-Rg1的吸收没有显著影响,提示G-Rg1可能不是P-gp的底物。
G-Rh1在Caco-2细胞单层模型双向转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良[25]。G-F1在Caco-2细胞单层模型,从AP→BL转运的Papp值在10-6~10-7数量级,预测其经肠道吸收中等或不良;从BL→AP转运的Papp值在10-5~10-6数量级[25]。
原人参三醇型皂苷的体内生物转化产物PPT在Caco-2细胞单层模型双向转运的Papp值在10-5~10-6数量级,预测其经肠道吸收良好[25]。
值得注意的是,某些原人参三醇型皂苷如G-Re本身对P-gp无抑制作用,但其肠内菌转化产物PPT是P-gp的抑制剂,与P-gp基质药物联合口服用药可能存在药物相互作用的风险,需慎重[36]。另一方面,与P-gp抑制剂合用,能增加人参皂苷的吸收。如在L-MDR1细胞模型,与P-gp抑制剂五味子木脂素合用,能明显提高G-Rg2的摄取率[38]。
2.2在体实验
将G-Rb1给大鼠灌胃、十二指肠和门静脉注射相对于尾静脉注射的绝对生物利用度分别为0.64%、2.46%、59.49%,与胃和肝消除相比,存在明显的肠首过作用[26]。给糖尿病大鼠和正常大鼠及高脂饲喂大鼠灌胃G-Rb1,生物利用度分别为2.25%、0.90%和0.78%,此可能与G-Rb1能促进糖尿病大鼠肠细胞间通透性有关[27]。按200mg·kg-1剂量灌胃给予大鼠G-Rb1,给药后7h或15h,在无菌大鼠血浆中既检测不到G-CK,亦检测不到其他的代谢产物,给予的G-Rb1大部分从肠道和累积粪便,特别是从盲肠中回收,证明其吸收性差;在无菌大鼠肠道定植真杆菌sp.A-44获得的单菌悉生大鼠,在血浆中可检测到相当量的G-CK,亦发现在盲肠和累积粪便中,给药后7h,在盲肠内容物中仅检测到少量的原形化合物G-Rb1。说明尽管G-Rb1肠道吸收性差,但它的肠内菌转化产物G-CK在肠道较下部分被吸收[5]。按5、10、20mg·kg-1剂量给大鼠灌胃G-CK,24h内在全部胃肠道中的原形G-CK回收率分别为54.3%、81.7%、77.5%,绝对生物利用度分别为1.8%、4.3%、35.0%,与低剂量相比,20mg·kg-1时的单位剂量AUC增加,绝对口服生物利用度也增加了[35]。但如果将G-CK在肝内形成的脂肪酸酯化物(G-CK-EM1)[40]计算在内,其生物利用度还要高。按每小鼠2mg的剂量灌胃G-Rb1后24h,在血清中未检测到原形化合物;给药后8h,血清中G-CK-M1达峰值(8.5±0.4)μg·mL-1,而直接给予G-CK-M1后2h,血清中G-CK-M1达峰值(10.3±1.0)μg·mL-1[40]。志愿受试者口服人参粉12g,服用后2h在血浆中即可检测到G-CK,其最高吸收发生在给药后9~14h,某些受试者甚至在给药后24h在血浆中仍可检测到G-CK[41]。
志愿受试者口服人参提取物或给大鼠灌胃人参总皂苷,在血浆中可检测到代谢产物G-CK、3-O-二去葡萄糖基G-Rb2(3-O-dideglucosylginsenoside Rb2)、G-C-Mc(ginsenoside C-Mc)和20(S)-PPT[13],提示某些人参皂苷以代谢产物的形式吸收入血。
给大鼠灌胃三七提取物,G-Rb1的生物利用度为4.35%[42];灌胃三七总皂苷,G-Rb1的生物利用度为1.18%[43]。G-Rd在犬的口服生物利用度为0.26%[44];给大鼠灌胃三七总皂苷,G-Rd的绝对生物利用度为2.36%[43]。G-Rg3在大鼠的口服生物利用度为2.63%[45]。G-Rh2在大鼠和犬的口服生物利用度分别为5%和16%[46]。G-Re在小鼠的口服绝对生物利用度为0.19~0.28%;在相对于纯G-Re等剂量给药,人参浆果提取物中的G-Re在小鼠的口服绝对生物利用度为0.33~0.75%[47],提示人参浆果提取物中的其他成分有促进G-Re吸收的作用。给大鼠灌胃三七总皂苷,G-Re的绝对生物利用度为7.06%[43]。
G-Rg1在大鼠的口服生物利用度为1.33%[48]。给大鼠灌胃三七提取物,G-Rg1的生物利用度为18.40%[42];灌胃三七总皂苷,G-Rg1的绝对生物利用度为6.06%[43]。
按10mg·kg-1剂量给大鼠灌胃G-Rg2,在血浆中检测不到原形化合物[49]。G-Rh1在大鼠的口服绝对生物利用度为(1.01±0.03)%[50],生物利用度较低。给大鼠灌胃三七总皂苷,NG-R1的绝对生物利用度为9.29%[43]。
将三七总皂苷给大鼠灌胃、十二指肠和门静脉注射相对于尾静脉注射的绝对生物利用度分别为3.29%、6.60%、50.56%,与胃和肝消除相比,存在明显的肠首过作用[27]。
20(R)-达玛烷-3β,12β,20,25-四醇[20(R)-dammarane-3β,12β,20,25-tetrol]在大鼠的绝对生物利用度为(64.8±14.3)%[51]。
PPD型人参皂苷(G-Ra3、Rb1、Rd、Rg3、Rh2)和PPT型人参皂苷(G-Rg1、Re、Rh1和NG-R1)的口服生物利用度一般小于5%,PPT型人参皂苷有稍好的生物利用度可能是因为其降解比PPD型人参皂苷慢[52]。
中药配伍应用可提高人参皂苷的生物利用度,如“生脉散”中的五味子木脂素能提高人参皂苷(如G-Rb1、Rb2、Rc、Rd、Rg2、Rg3等)的体内暴露量[38]。
传统上认为人参皂苷的吸收性差,生物利用度低。但如果考虑到人参皂苷的肠内细菌所致生物转化产物的吸收,如生物活性物质G-CK的吸收,则人参皂苷的吸收并非很差,则会得到不同的结论。
3 分布
有关人参皂苷体内分布的研究相对较少。在大鼠或小鼠,按20、50、150mg·kg-1剂量血管内给予G-Rd,小鼠在给药后0.5~72h,大鼠在给药后0.5~48h,在心、肝、脾、肺、肾、胃、肠、睾丸、子宫、脑、肌肉和脂肪组织等均可检测到G-Rd,其中以肺中分布量最高[53]。
给小鼠静脉注射G-CK后15min,在肝脏和小肠上部分布大量的G-CK,肺和膀胱较少,肾脏很难检测到,说明G-CK存在肠肝循环。按25mg·kg-1剂量给小鼠静脉注射G-CK后40min,G-CK的分布仍以肝脏和小肠为最高,血液和肺及肾脏最低,占给药剂量的50%。按25mg·kg-1剂量给小鼠一次静脉注射G-CK,肝脏中最高G-CK浓度出现在给药后10min,占给药剂量的65%。口服给予G-CK,依次通过胃、小肠、盲肠和结肠,其在胃肠道的分布量与G-CK的移行密切相关;口服给药后2h在肝脏中达最高,占给药量的8%,然后缓慢降低,说明G-CK自小肠吸收,聚积在肝脏[54]。
BST204是一个纯化的人参干燥提取物,含有高浓度的G-Rh2和Rg3差向异构体混合物[20(S)-G-Rh2、20(R)-G-Rh2、20(S)-G-Rg3、20(R)-G-Rg3],已用于癌症患者。按2g·kg-1剂量给大鼠灌胃BST204后120、240min,在脂肪组织、心、肾、肝、肺、肌肉、脾、胃、肠系膜、大肠和小肠皆可检测到20(S)-G-Rh2和20(S)-G-Rg3,小肠中分布量最高,其次为胃、肠系膜、肝和大肠,在脑组织中没有检测到20(S)-G-Rh2,但可检测到20(S)-G-Rg3;相同组织分布的20(S)-G-Rh2和20(S)-G-Rg3的量有所差异。可能由于20(R)-G-Rh2和20(R)-G-Rg3的吸收性差,在这些组织中皆没有检测到,此与血浆中的现象拟合良好[55]。
给大鼠静脉注射20(R)-G-Rg3,其在肝中分布最高,脑中分布最低;小肠壁、胃壁、肺、肾、脾中分布较高,心、脂肪、肌肉、卵巢、睾丸中的分布较低;在大鼠体内消除较快,其在各主要组织中的分布在给药后8h均显著降低[56]。
给家兔眼涂抹20(S)-G-Rg3眼膏后,房水、角膜和虹膜-睫状体中20(S)-G-Rg3浓度达到高峰的时间分别为0.42、0.16、0.19h[57]。给大鼠按1.5mg·kg-1剂量肌注20(S)-G-Rg3,在心、肝、脾、肺、肾中可以检测到20(S)-人参皂苷Rg3存在,而在脑、胃、肠、体脂、肌肉和生殖腺中均未检测到[58]。
按3mg·kg-1剂量给大鼠灌胃20(R)-G-Rh2后2、6h,在心、肝、脾、肺、肾、胃(不含内容物)、肠(不含内容物)、脂肪组织、卵巢皆可检测到20(R)-G-Rh2。此与血浆中的现象拟合良好[46]。给药后12h,除在上述所有组织外,还在肾上腺检测到了20(R)-G-Rh2。但在脑、皮肤、肌肉和睾丸没有检测到20(R)-G-Rh2。
给大鼠按50mg·kg-1剂量灌胃G-Rg1后8h,肝脏中浓度达到峰值,同时在脑核(包括皮层、海马、纹状体)、心、脾、肺、肾、胃、小肠、肌肉、脂肪中均有分布,24h后除肝脏外,上述其他组织中仍有G-Rg1存在,提示G-Rg1能透过血脑屏障进入脑部,这也很好地解释了其对中枢神经系统的促进学习记忆作用[59]。按100mg·kg-1剂量给大鼠灌胃G-Rg1,给药后150min处死动物,在肝、肾、心、肺、脾、胃、小肠、大肠、血清中皆可检测到G-Rg1。大鼠组织或血清中G-Rg1的浓度低于10μg·g-1或10μg·mL-1;消化道中G-Rg1是给予剂量的(77.3±3.9)%。Feng等[60]按10mg·kg-1剂量给大鼠静脉注射G-Rg1,给药后5、15、30min和1.0、1.5、2.0、3.0、4.0、6.0、8.0、2.0、18.0、24.0h取组织分析了G-Rg1的组织分布动力学特征,在心、肝、脾、肺、肾、脑、胰腺皆可检测到G-Rg1,同时在这些组织检测了G-Rg1的代谢产物G-Rh1和G-F1。除在脑中没有检测到G-Rh1和G-F1外,在上述组织中皆检测到了这2个代谢产物,原形化合物G-Rg1及其代谢产物G-Rh1和G-F1在上述组织分布良好,给药后5min,G-Rg1即可达到最高浓度,它在组织中的清除除肾外,皆比它在血中的清除快;代谢产物G-Rh1和G-F1在给药后4~6h即可达到最高浓度。刘梅等[61]探讨了G-Rg1经PEG修饰后在小鼠的组织靶向分布。分别给予小鼠尾静脉注射G-Rg1和PEG G-Rg1,于不同时间点取血并取各组织脏器,采用UPLC对样品中指标性成分G-Rg1进行含量测定,计算G-Rg1在各组织分布的靶向系数,观察比较2组在小鼠体内的组织靶向分布情况。结果G-Rg1组在小鼠体内各组织分布的AUC大小顺序为肝、肾、肺、心、脾,肝靶向系数为2.01;PEG修饰组在小鼠体内各组织的AUC大小顺序为肾、肝、肺、心、脾,肝靶向系数为9.21。说明PEG修饰后,G-Rg1对肝组织的靶向选择性增强,同时对肾、肺组织的靶向选择性亦得到增强。
已有报道新人参二醇(neoginsenoside;NPD;dammar-20(22)E-ene-3β,12β,25-triol)对神经具有保护作用。按100mg·kg-1剂量给大鼠灌胃NPD,在胃内容物、胃壁、肠内容物、大肠、肾、肝、胰腺、肺、小肠、肌肉、睾丸、脑、子宫、腹壁平滑肌、体脂肪、脾、卵巢、心、十二指肠皆可检测到NPD;给药后1.5~3.5h分布量达最高,24h已检测不到NPD,分布量最高的是胃和肠,其次为肾和肝,说明NPD在大鼠组织分布迅速,不会长期蓄积[62]。
20(R)-达玛烷-3β,12β,20,25-四醇[20(R)-dammarane-3β,12β,20,25-tetrol]对人胰腺癌细胞系HPAC和Panc-1细胞显示出比G-Rg3和PPD更强的细胞毒活性(IC50=~10μg·mL-1);按10mg·kg-1·d-1剂量、每周5d、连续腹腔给药6周,能够抑制无胸腺异种移植人胰腺癌小鼠肿瘤的生长,抑制率达52%。按10和20mg·kg-1剂量无论是静脉注射还是灌胃给予异种移植人胰腺肿瘤裸鼠20(R)-达玛烷-3β,12β,20,25-四醇,可分布于肿瘤部位、脾、肝和肾,肝中分布量最高[63]。
4 代谢和药代动力学
关于人参皂苷的代谢研究,主要围绕的是其在肠内的生物转化,而在体内的代谢研究相对比较少。在诸多的研究成果中,十分有意义的是人参二醇组皂苷,如G-Rb1、Rb2、Rc、Rd等的肠内菌转化产物G-CK。G-CK一部分随粪便排出体外,一部分吸收进入血液循环。如果将G-CK直接灌胃或静脉给予小鼠,在肝脏均可检出G-CK和它的代谢产物脂肪酸酯G-CK-EM1(EM1)[40,54]。而且静脉给药在小肠内除检出G-CK外,既没有检出EM1亦没有检出其他的代谢产物。给大鼠经尾静脉注射G-CK,EM1在肝脏的产率为给予剂量的24mol%,G-CK的回收率为给予剂量的30mol%。说明EM1在肝脏具有选择性滞留;EM1亦可能是在肝脏中合成的。从化学的角度分析,G-CK有亲脂性的功能团—达玛烷二醇基,这类似于胆甾醇;有亲水性的功能团—葡萄糖残基。已有研究证明:肝细胞通过受体(除半乳糖受体)识别葡萄糖。肝细胞的这种特异性功能决定了G-CK必定选择性在肝脏累积。G-CK通过肝脏从血液转运至胆汁,实际上是G-CK在肝细胞的内流和外流反应。EM1的产生,证明了G-CK的再代谢或转化。酯化反应可能类似于胆固醇与脂肪酸的酯化反应,由乙酰辅酶A催化完成。G-CK从肝脏被排入胆汁而进入肠道,可能存在肠肝循环。从EM1的FAB-MS数据、酸和碱水解分析,推断一分子EM1结合一分子脂肪酸;脂肪酸的种类主要为油酸和硬脂酸,少量为棕榈酸;脂肪酸残基结合的位点可能是C3-OH,也可能是C20-葡萄糖残基的C6-OH。与G-CK相比,EM1在肝脏中的滞留时间大大加长;G-CK可通过胆汁分泌进入小肠,而EM1则不能进入小肠。G-CK的肝脏代谢如图1所示。
G-CK对肿瘤细胞增殖具有抑制作用,攻击肿瘤细胞的第一个靶点是细胞膜。G-CK与血浆细胞膜相互作用,引起葡萄糖流入肿瘤细胞降低,抑制药物从肿瘤细胞外流。将G-CK与肿瘤细胞温孵培养,15min内,G-CK进入胞液和细胞核,通过上调细胞周期蛋白依赖性激酶抑制剂p27Kipl、下调c-Myc和细胞周期蛋白D1、活化半胱天冬酶-3活性,最终导致细胞分裂周期被阻断、细胞凋亡。G-CK及其EM1都能抑制B16-F10黑色素瘤细胞的生长,但EM1的毒性比G-CK低。在整体试验中,将EM1和G-CK分别直接注入(静脉注射可能会导致血浆酯酶水解EM1)肝移植性B16-F10黑色素瘤细胞癌灶内,发现EM1对肿瘤细胞生长的抑制作用比G-CK更强;在抗癌细胞转移方面,两者的作用几乎相同。
给大鼠按1、2、10mg·kg-1剂量静脉注射G-CK,AUC与剂量成正比,10mg·kg-1剂量的PK参数:半衰期(t1/2)=(224±115)min,AUC=(486.8±188.2)μg·min·mL-1,平均滞留时间(MRT)=(79.9±40.5)min,清除率(Cl)=(23.1±8.4)mL·min-1·kg-1;给大鼠按5、10、20mg-1剂量灌胃给予G-CK,AUC与剂量成正比,20mg-1剂量的PK参数:达峰时间(tmax)=(151±122)min,达峰浓度(Cmax)=(0.726±0.386)μg·mL-1,AUC=(341.0±201.8)μg·min·mL-1[35]。志愿受试者口服人参粉12g,G-CK血浆中的平均tmax、Cmax,AUC分别为(10.76±2.07)h、(27.89±24.46)ng·mL-1、(221.98±221.42)μg·h·mL-1[41]。志愿受试者口服人参提取物,在血浆中可检测到代谢产物G-CK、3-O-二去葡萄糖基G-Rb2(3-O-dideglucosylginsenoside Rb2)、G-C-Mc(ginsenoside C-Mc)和20(S)-PPT,血药浓度为0.3~5.1μg·[13]。
按400mg·kg-1剂量给大鼠灌胃G-Rb1,从尿液中得到8个代谢产物,分别为G-Rd、GY-XVII、G-F2、G-CK、3-酮基-G-CK、25-羟基-20(22)E-烯-G-Rg3、20(22),24-二烯-G-Rg3、25-羟基-G-Rd[64]。根据代谢产物化学结构特点,推测其可能的代谢途径为:G-Rb1→G-Rd→G-F2→G-CK→3-酮基-G-CK或G-Rb1→G-Rd→20(S)-G-Rg3→20(22)E,24-二烯-G-Rg3→25-羟基-20(22)E-烯-G-Rg3或G-Rb1→G-Rd→20(R)-G-Rg3(或25-羟基-G-Rd)或G-Rb1→GY-XVII→G-F2→G-CK→3-酮基-G-CK。
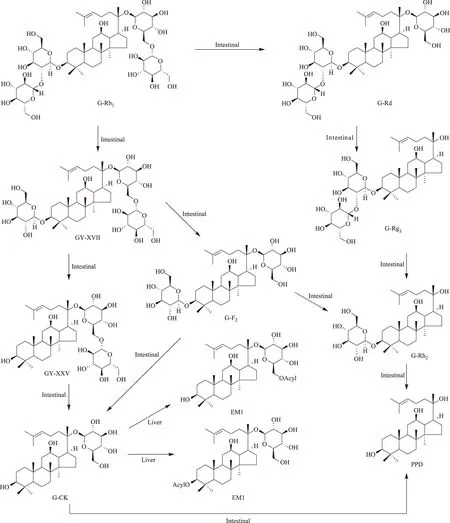
图1 G-Rb1的生物转化和G-CK的肝脏代谢途径
按50、100、200mg·kg-13个剂量给大鼠灌胃G-Rb1,有其药动学特征,t1/2分别为30.42、19.38、31.34h,tmax分别为8、6、6h,AUC0→t分别为343.17、565.18、7439.79mg·L-1·h-1,AUC0→∞分别为506.34、706.47、12165.36mg·L-1·h-1,Vd分别为1.734、0.792、0.074L·kg-1,Cl分别为0.039、0.028、0.002L·kg-1,MRT0→t分别为17.88、17.802、20.05h,MRT0→∞分别为41.73、29.43、21.58h,ρmax分别为18.03、34.31、375.66μg·L-1[65]。说明尽管G-Rb1口服生物利用度差,但仍有吸收,并具有一定的药动学特征。
按0.4mg·kg-1剂量给大鼠静脉注射G-Rc,在尿液中可检测到原形G-Rc及其连续脱去C-3吡喃葡萄糖基的代谢产物G-Rc-Mb和G-Rc-Mc,而灌胃给药,在粪便中可检测到原形G-Rc,大部分的G-Rc代谢为G-Rc-Mc和G-CK。大鼠静脉注射G-Rc的血药浓度-时间PK为二房室模型,主要PK参数为:t1/2α=(7.30±1.13)min,t1/2β=(1091.67±173.18)min,k10=(0.201±0.004)·h-1,k12=(4.49±0.57)·h-1,k21=(1.12±0.32)·h-1,AUC=(1701.19±144.81)μg·min·mL-1,Vc=(23.97±2.52)mL,Cl=(4.81±0.41)mL·h-1[66]。
灌胃和静脉给予大鼠G-Rd,利用LC-MS方法检测,比较给药后0~24h内收集的尿中的代谢产物,发现两种给药途径下的代谢产物有较大差异。灌胃给药后的代谢反应包括糖苷键的水解反应和结合反应,静脉给药后药物的代谢转化以氧化反应和结合反应为主。灌胃给药后的尿液中检测到4个代谢产物,静脉给药后的尿液中检测到了5个代谢产物,它们为G-Rd的单氧化产物、G-Rd、G-Rg3和G-Rh2等[67-68]。说明G-Rd在给药24h后,大鼠体内仍有原形存在。文中叙述代谢产物中还有G-Rb1存在[68],产生机制不明。
按50、100、200mg·kg-13个剂量组给大鼠灌胃G-Rd,有其药动学特征,t1/2分别为11.09、17.76、9.83h,tmax分别为8、8、8h,AUC0→t分别为223.70、1109.12、9682.74mg·L-1·h-1,AUC0→∞分别为240.56、1241.10、10108.36mg·L-1·h-1,Vd分别为0.078、0.016、0.002L·kg-1,Cl分别为1.331、0.413、0.028L·kg-1,MRT0→t分别为15.08、15.80、17.97h,MRT0→∞分别为18.51、21.95、19.92h,ρmax分别为26.45、118.31、866.35μg·L-1[44]。按20、50、150mg·kg-1单剂量给小鼠静脉注射G-Rd,血药浓度-时间曲线的PK特征为开放二房室模型,20mg·kg-1时,分布半衰期t1/2α=(0.21±0.11)h,清除半衰期t1/2β=(14.19±2.37)h,中央隔室的表观体积Vc=(0.375±0.101)L,Cl=(0.066±0.005)·h-1,AUC=(305.0±22.3)μg·mL-1·h-1;50mg·kg-1时,t1/2α=(0.66±0.69)h,t1/2β=(12.83±2.92)h,Vc=(0.809±0.565)L,Cl=(0.280±0.172)·h-1,AUC=(293.2±279.4)μg·mL-1·h-1;150mg·kg-1时,t1/2α=(0.48±0.25)h,t1/2β=(14.02±10.57)h,Vc=(1.432±0.242)L,Cl=(0.569±0.306)·h-1,AUC=(312.6±139.5)μg·mL-1·h-1[53]。3个剂量下,给药后较早的2min,血药浓度即达峰值,然后迅速下降;至给药后1h,迅速下降至近70%;给药后8~24h,降低到给药剂量的90%以上,类似的趋势亦呈现在大鼠。给予犬按0.2mg·kg-1剂量静注和2mg·kg-1剂量灌胃G-Rd,静注时t1/2=(39.4±12.0)h,MRT=(26.7±1.63)h,AUC0→t=(76403.4±15880.6)ng·h·mL-1,AUC0→∞=(76451.1±15874.8)ng·h·mL-1,Cl=(0.002±0.0005)L·kg-1;灌胃时t1/2=(24.2±2.85)h,MRT=(25.5±3.84)h,AUC0→t=(1890.2±668.6)ng·h·mL-1,AUC0→∞=(1930.3±647.4)ng·h·mL-1,Cl=(1.14±0.40)L·kg-1[44];按10mg剂量给志愿受试者静脉滴注G-Rd,Cmax=(2841.18±473.03)ng·mL-1,tmax=(0.50±0.00)h,t1/2=(19.29±3.44)h,AUC0→t=(27261.63±8116.88)ng·h·mL-1,AUC0→∞=(27929.39±8615.75)ng·h·mL-1,MRT0→t=(17.52±3.73)h,MRT0→∞=(19.79±5.24)h,Vd=(10.57±2.88)L·kg-1,Cl=(0.39±0.12)L·h-1[68],静注G-Rd,在人体内代谢缓慢。
给大鼠按1.5mg·kg-1剂量肌注20(S)-G-Rg3,与血浆蛋白的结合率为15%~17%,且不随浓度的变化而改变。PK行为符合一室模型,t1/2(ke)=0.75h,tmax=0.116h,ρmax=13.9mg·L-1,AUC=16.6mg·h·L-1[58]。提示肌内注射后很快被吸收进入血液,迅速达到血药浓度峰值,在体内的代谢速度亦较快。家兔涂抹20(S)-G-Rg3眼膏后,其在房水、角膜和虹膜-睫状体中的PK行为均符合一室模型,房水、角膜和虹膜-睫状体中tmax分别为0.42、0.16、0.19h,t1/2分别为0.71、0.69、0.78h,3h后眼组织中的药物浓度已低于最大浓度的1/10[57]。
相对于20(S)-G-Rg3,文献中对20(R)-G-Rg3的代谢和PK报道较多,是由于化学结构画错,还真的就是20(R)-G-Rg3的代谢和PK结果,有待于查证。按100mg·kg-1剂量给大鼠灌胃20(R)-G-Rg3,血浆中没有检测到原形化合物,给药后24h累积粪便中原形化合物的回收率仅是给予剂量的0.97%~1.15%,提示其发生了快速、广泛的生物转化和/或代谢,代谢产物包括20(R)-G-Rh2、PPT、PPT单氧化物、PPT二氧化物、20(R)-G-Rg3单氧化物和20(R)-G-Rg3二氧化物等。按5mg·kg-1剂量静脉给药,t1/2仅为18.5min,给药后1.5h全部消除[69]。另有报道[56],分别以0.25、0.5、1.0mg·kg-1剂量给大鼠静脉注射20(R)-G-Rg3,其药动学参数分别为:t1/2=(0.62±0.10)、(0.61±0.08)和(0.62±0.08)h,AUC0→t=(145±27.9)、(302±58.2)和(698±128)ng·mL-1·h-1,AUC0→∞=(147±28.7)、(304±57.9)和(700±128)ng·mL-1·h-1,MRT=(0.17±0.03)、(0.13±0.03)和(0.16±0.04)h,Cl=(1.43±0.32)、(1.42±0.32)和(1.31±0.22)mL·min-1·kg-1,Vss=(29.3±5.57)L·kg-1、(28.3±5.44)和(27.4±6.52)L·kg-1。AUC与剂量成正比。给beagle犬口服(2mg·kg-1)或静脉注射(0.3mg·kg-1)20(R)-G-Rg3,在血浆中没有检测到代谢产物20(R)-G-Rh2和PPT[70];主要PK参数,口服给药Cmax=(7.30±1.67)ng·mL-1,tmax=(2.50±0.84)h,t1/2=(5.99±1.16)h,ke=(0.12±0.02)·h-1,AUC0→t=(45.9±12.8)ng·h·mL-1,AUC0→1=(50.0±13.1)ng·h·mL-1,MRT=(6.55±1.40)h,Vd=(722±257)L·kg-1,Cl=(362±91.4)mL·min-1·kg-1;静脉给予t1/2=(1.71±0.11)h,ke=(0.42±0.05)·h-1,AUC0→t=(1458±437)ng·h·mL-1,AUC0→∞=(1466±444)ng·h·mL-1,MRT=(0.85±0.18)h,Vd=L·kg-1,Cl=(3.65±0.99)mL·min-1·kg-1。按10mg·kg-1剂量给大鼠灌胃20(R)-G-Rg3,血浆中可检测到原形化合物及其代谢产物20(R)-G-Rh2和PPT;20(R)-G-Rg3的Cmax=(104.07±59.95)ng·mL-1,tmax=(4.40±1.67)h;按1mg·kg-1剂量给大鼠静脉注射20(R)-G-Rg3,其血药浓度-时间PK特征为二房室模型,t1/2α=(0.12±0.03)h,t1/2β=(2.09±0.50)h[45]。志愿受试者按3.2mg·kg-1单剂量口服20(R)-G-Rg3,Cmax=(15.67±6.14)ng·mL-1,tmax=(0.66±0.01)h[71]。6名健康志愿者按0.8mg·kg-1单剂量口服20(R)-GRg3,由于血药浓度低,可测数据点少,未进行模型模拟;说明口服G-Rg3后在体内吸收快,消除也较快,但血药浓度很低。8名健康志愿者按3.2mg·kg-1单剂量口服20(R)-G-Rg3,其药时曲线符合口服吸收有滞后时间的二房室模型,tmax=(0.66±0.10)h,Cmax=(16±6)ng·mL-1,t1/2α=(0.46±0.12)h,t1/2β=(4.9±1.1)h,t1/2(Ka)=(0.28±0.04)h,AUC0→∞=(77±26)ng·mL-1·h-1[72]。
给大鼠按400、1000、2000mg·kg-1单剂量灌胃含有20(S)-G-Rh2、20(R)-G-Rh2、20(S)-G-Rg3、20(R)-G-Rg3的纯化人参干燥提取物BST204,在血浆中仅能检测到20(S)-G-Rh2和20(S)-G-Rg3,而检测不到20(R)-G-Rh2和20(R)-G-Rg3[55];AUC0→t和Cmax与剂量成正比;但终末t1/2和tmax略有不同,在3个剂量下,20(S)-G-Rh2的t1/2分别为(161±88.3)、(145±131)、(140±50.3)min,tmax分别为90(60~120)、120(120~240)、90(60~120)min;20(S)-G-Rg3的t1/2分别为(147±25.8)、(304±40.0)、(406±181)min,tmax分别为270(240~600)、240(120~480)、270(120~300)min。纯品20(S)-G-Rh2(31.5mg·kg-1)和20(S)-G-Rg3(68.0mg·kg-1)分别给予雄性大鼠灌胃,20(S)-G-Rh2的PK参数:AUC0→t=(234±110)g·min·mL-1,终末t1/2=(172±105)min,Cmax=(1.26±0.577)g·mL-1,tmax=120(90-240)min,GI24剂量百分数(%)=(1.08±1.57)h,Ae24剂量百分数(%)=(1.01±1.23)h;20(S)-G-Rg3的PK参数:AUC0→t=(107±66.6)g·min·mL-1,终末t1/2=(241±73.0)min,Cmax=(0.280±0.221)g·mL-1,tmax=300(240-480)min,GI24剂量百分数(%)=(1.24±1.85)h,Ae24剂量百分数(%)=(2.40±2.38)h。提示纯品和混合物形式给药PK行为不同。
给大鼠按0.03、0.1、0.3mg·kg-1单剂量静脉注射20(R)-G-Rh2,PK参数,Cmax分别为288、683、1069ng·mL-1,t1/2分别为1.8、4.9、4.5h,MRT分别为0.8、2.4、1.9h,Cl分别为15.2、20.9、35.4mL·min-1·kg-1,Vd分别为2.3、8.9、13.7L·kg-1,AUC0→t分别为32.1、75.0、134.5ng·h·mL-1,AUC0→∞分别为32.9、79.8、141.4ng·h·mL-1;给大鼠按1、3、9mg·kg-1单剂量灌胃20(R)-G-Rh2,tmax分别为6、6、6h,Cmax分别为3.5、8.7、65.3ng·mL-1,t1/2分别为4.0、3.9、2.6h,MRT分别为9.6、9.3、7.1h,Cl分别为20.9、20.9、20.9mL·min-1·kg-1,Vd分别为7.3、7.1、4.7L·kg-1,AUC0→t分别为35.1、93.4、457.1ng·h·mL-1,AUC0→∞分别为37.1、95.8、460.2ng·h·mL-1,F分别为4.7%、4.0%、6.4%;20(R)-G-Rh2与大鼠血浆蛋白的结合率为70%[46]。给Beagle犬按0.1mg·kg-1剂量单次静脉注射20(R)-G-Rh2,Cmax=(1735.8±266.4)ng·mL-1,t1/2=(7.8±0.9)h,MRT=(4.0±0.5)h,Cl=(2.1±0.5)mL·min-1·kg-1,Vd=(1.4±0.4)L·kg-1,AUC0→t=(813.6±196.7)ng·h·mL-1,AUC0→∞=(844.6±202.3)ng·h·mL-1,F=100%;给Beagle犬按1mg·kg-1剂量单次口服20(R)-G-Rh2,tmax=(2.6±1.3)h,Cmax=(371.0±199.6)ng·mL-1,t1/2=(9.6±3.5)h,MRT=(6.4±2.4)h,Cl=(2.1±0.5)mL·min-1·kg-1,Vd=(1.8±1.0)L·kg-1,AUC0→t=(1190.2±610.0)ng·h·mL-1,AUC0→∞=(1243.2±622.3)ng·h·mL-1,F=(16.7±11.7)%[46]。Beagle犬静脉给药的血药浓度-时间PK特征为三室模型,说明20(R)-G-Rh2嗣后缓慢地再分布到周边室;多次给药不影响其PK特征。在大鼠灌胃20(R)-G-Rh2剂量3mg·kg-1组的给药后0~24h累积粪便中检测到代谢产物PPT,胆汁中亦存在20(R)-G-Rh2的代谢产物,尿液中检出20(R)-G-Rh2的硫酸酯共价结合物。
给大鼠静脉注射G-Re,收集药后0~72h的尿液,可发现原形化合物和9个代谢产物,包括3个G-Re的单加氧产物,主要代谢产物20(S)-G-Rg1以及20(S)-G-Rg2、20(R)-G-Rg2、G-Rh1或G-F1、PPT、去氢PPT。由于是静脉给药,证明这些代谢产物在体内发生。给大鼠灌胃G-Re,收集药后0~72h的尿液,可发现原形化合物和8个代谢产物,20(S)-G-Rg1、20(S)-G-Rg2、20(R)-G-Rg2、G-Rh1或G-F1、PPT、去氢PPT,以及2个未确定结构的代谢产物[73]。主要代谢途径为氧化和脱糖基。
给ICR小鼠按1mg·kg-1剂量静脉给予G-Re,PK参数,在雄性小鼠AUC0→t=(638.8±197.0)ng·h·mL-1,AUC0→∞=(639.3±196.8)ng·h·mL-1,t1/2=(0.2±0.03)h,MRT=(0.2±0.07)h,Vd=(0.3±0.2)L·kg-1,Cl=(1.7±0.7)L·h-1·kg-1;在雌性小鼠AUC0→t=(1437.6±271.2)ng·h·mL-1,AUC0→∞=(1442.0±271.0)ng·h·mL-1,t1/2=(0.5±0.08)h,MRT=(0.5±0.08)h,Vd=(0.2±0.07)L·kg-1,Cl=(0.7±0.11)L·h-1·kg-1[47],存在性别差异。单体化合物G-Re与人参浆果提取物(含等量G-Re)给药相比,灌胃给药,10mg·kg-1剂量组,单体化合物G-Re的PK参数,tmax=(0.4±0.2)h,Cmax=(29.0±25.4)ng·mL-1,AUC0→t=(17.7±4.5)ng·h·mL-1,MRT=(0.76±0.20)h,F=0.28%;人参浆果提取物组tmax=(0.3±0.13)h,Cmax=(21.3±10.1)ng·mL-1,AUC0→t=(21.3±19.9)ng·h·mL-1,MRT=(1.06±0.56)h,F=0.33%。50mg·kg-1剂量组,单体化合物G-Re的PK参数,tmax=(0.7±0.7)h,Cmax=(35.0±4.3)ng·mL-1,AUC0→t=(61.5±37.0)ng·h·mL-1,MRT=(2.0±1.2)h,F=0.19%;人参浆果提取物组tmax=(0.5±0.3)h,Cmax=(124.1±127.9)ng·mL-1,AUC0→t=(238.3±64.2)ng·h·mL-1,MRT=(4.0±2.0)h,F=0.75%。50mg·kg-1剂量组的Cmax和AUC0→t存在差异,提取物(F=0.33%~0.75%)中的G-Re更易吸收,但总体上口服吸收差F=0.19%~0.28%[47]。
按100和200mg·kg-1剂量给大鼠灌胃G-Re[65],100mg·kg-1剂量组,t1/2=2.928h,tmax=8h,AUC0→t=19.75mg·L-1·h-1,AUC0→∞=19.85mg·L-1·h-1,Vd=4.257L·kg-1,Cl=1.008L·kg-1,MRT0→t=8.89h,MRT0→∞=8.986h,ρmax=2.42μg·L-1;200mg·kg-1剂量组,t1/2=4.971h,tmax=8h,AUC0→t=90.32mg·L-1·h-1,AUC0→∞=99.66mg·L-1·h-1,Vd=1.44L·kg-1,Cl=0.201L·kg-1,MRT0→t=9.83h,MRT0→∞=11.83h,ρmax=14.32μg·L-1。血药浓度与给予剂量成正相关。按20、30、40mg·kg-1剂量给大鼠静脉注射G-Re,t1/2α分别为6.505、6.817、4.499min,t1/2β分别为28.96、30.49、27.57min,AUC分别为599.31、1025.65、1415.7min·mg·L-1。雄性与雌性大鼠均按1mg·kg-1静脉注射G-Re,其t1/2分别为0.2、0.5h,口服绝对生物利用度仅为0.33%~0.75%[74]。但未见对代谢产物进行分析。因此,G-Re真实的生物利用度无法判断。
志愿受试者口服200mg G-Re片[75],G-Re代谢为G-Rg1、G-F1、G-Rh1和PPT,但没有检测到G-Rg2,提示G-Rg2可能在体内迅速被代谢。即或G-Re在肠内被转化为G-Rg1、G-F1、G-Rh1和PPT,能够在尿液中检测到这些产物,证明它们能够被吸收。G-Re在血液中的PK参数tmax=(1.19±0.44)h,Cmax=(0.939±0.549)μg·L-1,AUC0→t=(2.476±2.281)μg·L-1·h-1,AUC0→∞=(2.699±2.284)μg·L-1·h-1,t1/2=(1.82±0.75)h,Cl/F=(124054±84725)L·h-1。
G-Rg1在大鼠肠内可转化为G-Rh1、G-F1和PPT;在人肠内可转化为G-Rh1和PPT[60]。
按100和200mg·kg-1剂量给大鼠灌胃G-Rg1[65],100mg·kg-1剂量组,t1/2=7.725h,tmax=8h,AUC0→t=66.11mg·L-1·h-1,AUC0→∞=72.66mg·L-1·h-1,Vd=3.068L·kg-1,Cl=0.275L·kg-1,MRT0→t=13.52h,MRT0→∞=16.549h,ρmax=7.70μg·L-1;200mg·kg-1剂量组,t1/2=4.731h,tmax=8h,AUC0→t=284.73mg·L-1·h-1,AUC0→∞=288.42mg·L-1·h-1,Vd=0.473L·kg-1,Cl=0.069L·kg-1,MRT0→t=11.93h,MRT0→∞=12.321h,ρmax=38.38μg·L-1。与G-Re的药代动力学行为基本一致。按10mg·kg-1剂量给大鼠静脉注射G-Rg1,血药浓度-时间曲线PK特征为二房室模型[60],给药后5min,G-Rg1血药浓度最高,然后迅速下降,给药后4h消失;代谢产物G-Rh1和G-F1在给药后1h 在血中出现,约在6h 达到最高血药浓度,给药后24h在血中完全消失;但没有发现PPT。PK参数,G-Rg1的α=(2.2±0.8)·h-1,β=(0.28±0.11)·h-1,t1/2α=(0.37±0.13)h,t1/2β=(1.82±0.64)h,Vc=(0.021±0.009)L·kg-1,Cl=(0.037±0.017)mL·h-1·kg-1,MRT0→t=(1595.7±451.6)ng·h·mL-1,MRT=(1.92±0.23)h;G-Rh1的α=(2.9±1.1)·h-1,β=(0.16±0.04)·h-1,Cmax=(55.7±17.7)ng·mL-1,tmax=(5.32±0.63)h,t1/2α=(0.30±0.14)h,t1/2β=(5.87±2.66)h,Vc=(0.028±0.011)L·kg-1,Cl=(0.009±0.005)mL·h-1·kg-1,MRT0→t=(597.5±274.4)ng·h·mL-1,MRT=(5.99±3.03)h;G-F1的α=(4.1±1.3)·h-1,β=(0.13±0.05)·h-1,Cmax=(71.4±18.0)ng·mL-1,tmax=(6.88±0.86)h,t1/2α=(0.23±0.10)h,t1/2β=(6.87±2.95)h,Vc=(0.035±0.014)L·kg-1,Cl=(0.016±0.006)mL·h-1·kg-1,MRT0→t=(805.6±233.7)ng·h·mL-1,MRT=(7.13±2.25)h。
按150mg·kg-1剂量给大鼠灌胃G-Rg1,血中可检测到G-Rg1及其代谢产物G-Rh1、G-F1和PPT。血药浓度-时间曲线PK特征为二房室模型[60],给药后15min在血液中出现G-Rg1,给药后1h达最大血药浓度,然后迅速下降,给药后8h消失;代谢产物G-Rh1和G-F1约在给药后1h 在血中出现,约在4h达到平均最高血药浓度;代谢产物PPT约在给药后3h 在血中出现,约在8h达到最高血药浓度;给药后24h,仍可在血液中检测到三个代谢产物。PK参数,G-Rg1的α=(7.4±2.9)·h-1,β=(0.46±0.16)·h-1,Cmax=(1134.4±562.80)ng·mL-1,tmax=(0.92±0.12)h,t1/2α=(0.14±0.05)h,t1/2β=(2.25±0.68)h,Vc/F=(0.008±0.002)L·kg-1,Cl/F=(0.031±0.014)mL·h-1·kg-1,MRT0→t=(2363.5±914.80)ng·h·mL-1,MRT=(2.68±0.36)h。G-Rh1的α=(3.6±0.8)·h-1,β=(0.12±0.05)·h-1,Cmax=(584.6±117.3)ng·mL-1,tmax=(3.64±1.43)h,t1/2α=(0.29±0.09)h,t1/2β=(6.73±2.28)h,Vc/F=(0.015±0.040)L·kg-1,Cl/F=(0.017±0.080)mL·h-1·kg-1,MRT0→t=(4185.5±1364.2)ng·h·mL-1,MRT=(5.06±1.33)h。G-F1的α=(4.0±1.0)·h-1,β=(0.17±0.05)·h-1,Cmax=(522.2±197.5)ng·mL-1,tmax=(5.17±0.93)h,t1/2α=(0.22±0.08)h,t1/2β=(5.44±1.44)h,Vc/F=(0.020±0.007)L·kg-1,Cl/F=(0.022±0.005)mL·h-1·kg-1,MRT0→t=(3774.3±890.40)ng·h·mL-1,MRT=(6.65±2.17)h。PPT的α=(4.4±1.4)·h-1,β=(0.22±0.06)·h-1,Cmax=(38.3±23.4)ng·mL-1,tmax=(7.30±2.00)h,t1/2α=(0.20±0.04)h,t1/2β=(5.06±0.90)h,Vc/F=(0.027±0.011)L·kg-1,Cl/F=(0.011±0.006)mL·h-1·kg-1,MRT0→t=(396.2±202.6)ng·h·mL-1,MRT=(5.33±3.20)h。
人静脉注射20(S)-G-Rg1后,体内药动学参数t1/2α=(0.28±0.10)h,t1/2β=(2.09±1.89)h,VC=(0.015±0.006)L·kg-1,AUC0→t=(124.4±35.9)ng·L-1·h-1,AUC0→∞=(127.9±37.2)ng·L-1·h-1,Cl=(0.03±0.01)L·kg-1·h-1。提示20(S)-G-Rg1在人体内的分布与消除都较快[76]。
给大鼠静脉注射G-Rg2,血液中存在20(S)-G-Rg2和20(R)-G-Rg2。按10、20、50mg·kg-13个剂量分别单次静脉给药后,20(S)-G-Rg2具有代谢不稳定性,部分转化为20(R)-G-Rg2,两者的PK过程符合开放一房室模型,20(R)-G-Rg2在体内的消除半衰期较长,该对差向异构体其他主要药动学参数的差异具有统计学意义,可见两者的药动学是不同的[77]。
20(R)-G-Rg2的PK参数:A=(5.0005±0.0648)μg·mL-1,α=(0.1740±0.0220)min-1,B=(1.6259±0.1232)μg·mL-1,β=(0.0097±0.0004)min-1,t1/2α=(4.0246±0.0087)min,t1/2β=(71.1999±3.1586)min,k21=(0.0504±0.0065)min-1,k10=(0.0336±0.0003)min-1,k12=(0.0997±0.0157)min-1,Vc=(3.8115±0.0988)L·kg-1,Vd=(7.5398±0.5373)L·kg-1,AUC=(197.7176±5.1766)μg·min·mL-1,Cl=(0.1264±0.0003)L·min-1;20(S)-G-Rg2的PK参数:A=(55.3000±3.7722)μg·mL-1,α=(0.1963±0.0633)min-1,B=(14.5104±5.6855)μg·mL-1,β=(0.0181±0.0057)min-1,t1/2α=(3.7242±0.0459)min,t1/2β=(38.4414±1.1134)min,k21=(0.0562±0.0274)min-1,k10=(0.0640±0.0100)min-1,k12=(0.0942±0.0358)min-1,Vc=(0.3620±0.0459)L·kg-1,Vd=(0.6068±0.3821)L·kg-1,AUC=(1092.5109±83.9747)μg·min·mL-1,Cl=(0.0232±0.0013)L·min-1[78]。
给大鼠灌胃20(S)-G-Rh1,t1/2β=(0.43±0.08)h,tmax=(1.00±0.00)h,Cmax=(0.05±0.01)mg·L-1,AUC0→t=(0.13±0.01)mg·L-1·h-1,AUC0→∞=(0.14±0.01)mg·L-1·h,CL/F=(67.68±9.63)L·h-1;静脉注射,t1/2α=(0.07±0.04)h,t1/2β=(0.41±0.05)h,Vd=(0.13±0.04)L,AUC0→t=(1.50±0.23)mg·L-1·h-1,AUC0→∞=(1.52±0.24)mg·L-1·h-1,Cl=(0.67±0.11)L·h-1;F=(1.01±0.03)%。由此可见20(S)-G-Rh1在体内的清除较快,生物利用度较低[50]。
给大鼠按5mg·kg-1剂量静脉注射20(R)-达玛烷-3β,12β,20,25-四醇,血浆PK参数为t1/2=(3.9±2.1)h,ke=(0.2±0.1)h-1,AUC0→t=(31808±11685)ng·h·mL-1,AUC0→∞=(33487±14834)ng·h·mL-1,MRT=(4.7±2.9)h,Vd/F=(950.2±115.0)mL·kg-1,Cl/F=(166.6±52.4)mL·h-1·kg-1;按10mg·kg-1剂量灌胃给予20(R)-达玛烷-3β,12β,20,25-四醇,血浆PK参数为Cmax=(4617.2±1571.3)ng·mL-1,tmax=(5.5±4.7)h,t1/2=(4.5±2.6)h,ke=(0.2±0.1)h-1,AUC0→t=(38954±5172)ng·h·mL-1,AUC0→∞=(40194±6666)ng·h·mL-1,MRT=(8.1±3.1)h,Vd/F=(1355.4±510.6)mL·kg-1,Cl/F=(254.1±42.7)mL·h-1·kg-1[51]。给异种移植人胰腺肿瘤裸鼠,按10mg·kg-1剂量静脉注射20(R)-达玛烷-3β,12β,20,25-四醇,Cmax=41.9μg·mL-1,AUC0→∞=21.3μg·h·mL-1,t1/2=2.39h,MRT=1.03h,Cl/F=0.470mL·g-1·h-1;按20mg·kg-1剂量灌胃20(R)-达玛烷-3β,12β,20,25-四醇,Cmax=2.96μg·mL-1,AUC0→∞=5.36μg·h·mL-1,t1/2=1.52h,MRT=1.73h,Cl/F=3.73mL·g-1·h-1[63]。在大鼠的绝对生物利用度为64.8%[51];而在异种移植人胰腺肿瘤裸鼠的绝对生物利用度为19.8%[63]。在异种移植人胰腺肿瘤裸鼠,静脉注射20(R)-达玛烷-3β,12β,20,25-四醇在肿瘤部位的Cmax=4.36μg·g-1,AUC0→∞=128μg·h·g-1,t1/2=2.52h,MRT=2.28h;灌胃在肿瘤部位的Cmax=2.05μg·g-1,AUC0→∞=9.64μg·h·g-1,t1/2=3.21h,MRT=3.99h[63],在肿瘤部位的PK性质良好。
G-Rb1、Rb2、Rb3、Rc、Rd为PPD型皂苷,亦为人参的主要化学成分。PPD型皂苷在体内可代谢为PPD,PPT型皂苷在体内可代谢为PPT,因此,研究PPD和PPT的PK意义重大。给大鼠按75mg·kg-1剂量灌胃人参皂苷元(PPD和PPT分别占16%和33%)或按30mg·kg-1剂量静脉注射人参皂苷元,研究它们的PK特性。静脉给予,PPD的t1/2,λz=6.25h,Cl=0.98L·h-1·kg-1,清除相对较慢;PPT的t1/2,λz=0.80h,Cl=4.27L·h-1·kg-1,清除较快。灌胃给予,PPD和PPT皆可吸收进入体循环,PPD的tmax=1.82h,Cmax=1.04μg·mL-1,吸收相对较慢;PPT的tmax=0.58h,Cmax=0.13μg·mL-1,吸收相对较快。两者的系统暴露量有明显的差别。PPD和PPT的绝对生物利用度分别为48.12%和3.69%,差异明显[79]。这些差异性为研究PPD型皂苷和PPT型皂苷的作用,在药代动力学上奠定了基础。
给予动物或人单一的人参皂苷或人参皂苷混合物或人参皂苷提取物或含有人参/三七的复方,某些人参皂苷可能具有不同的PK行为。给大鼠静脉注射单一的G-Rb1、Rb2和Rb3,t1/2分别为(12.5±1.8)、(15.4±3.7)、(24.9±12.6)h;灌胃单一的G-Rb1、Rb2和Rb3,t1/2分别为(9.8±6.9)、(23.1±3.7)、(21.1±9.8)h[80]。给大鼠灌胃三七提取物,G-Ra3、Rb1、Rc、Rd的t1/2较长,为7.5~19.8h;而G-Rg1、Rg2、Rg3、F1、F2、Rh1、Rh2、Re、Rf,20-葡萄糖基G-Rf的t1/2相对较短,为0.2~3.2h[25]。同样是三七提取物,给大鼠灌胃的G-Rb1和Rg1的t1/2分别为17.96和14.13h[42]。给beagle犬灌胃三七提取物,G-Rb1、Rg1和NG-R1的t1/2分别为(18.27±2.55)、(4.55±1.22)和(3.35±0.55)h[81];给beagle犬灌胃含有三七提取物的丹参片,G-Rb1、Rg1和NG-R1的t1/2分别为(60.9±17.5)、(4.59±2.95)和(4.82±3.13)h[82];给大鼠灌胃七藿通窍片(三七提取物和淫羊藿提取物组成的复方中药制剂),G-Rb1、Re、Rg1和NG-R1的t1/2分别为(1512.2±1461.9)、(431.4±149.5)、(418.8±68.0)和(245.6±118.5)min[83];给大鼠静脉注射参附(红参和附子)注射液(剂量相当于G-Re,0.18mg·kg-1;G-Rf,0.10mg·kg-1;G-Rb1,0.66mg·kg-1;G-Rc,0.57mg·kg-1;G-Rb2,0.42mg·kg-1;G-Ro,0.08mg·kg-1;G-Rd,0.81mg·kg-1),Rb1、Rb2、Rc、Rd和G-Rf的t1/2分别为(19.29±6.36)、(35.60±30.66)、(29.54±22.91)、(8.49±5.20)和(4.21±3.68)h,AUC0→t分别为(71.37±31.17)、(44.42±20.24)、(65.30±29.10)、(2.81±2.02)、(4.38±3.30)mg·L-1·h-1,MRT0→t分别为(9.83±0.36)、(10.22±1.51)、(10.00±1.30)、(6.01±3.33)、(2.99±2.17)h[84]。在志愿受试者中[75],口服人参皂苷,G-Re的t1/2为(1.82±0.75)h;静脉注射参麦注射液[76],G-Rg1的t1/2为(2.09±1.89)h。同一人参皂苷在不同条件下的不同PK行为直接影响其生物学活性和药理学作用。
5 排泄
给大鼠灌胃G-Rb1,在尿液中可检测到原形化合物及其代谢产物G-Rd、Rg3、Rh2,说明G-Rb1可经尿液排泄[6]。
给大鼠静脉或灌胃给予G-CK,给药后24h在整个胃肠道的原形化合物的回收率分别为24.4%~26.2%和54.3%~81.7%,尿液中排泄量最少[35]。
志愿受试者口服人参提取物,在尿液中可检测到代谢产物G-CK、3-O-二去葡萄糖基G-Rb2(3-O-dideglucosylginsenoside Rb2)、G-C-Mc(ginsenoside C-Mc)和20(S)-PPT,尿药浓度为2.2~96μg·mL-1[13]。
给大鼠灌胃G-Rc,主要以一部分原形和代谢产物G-Rc-Mc和G-CK形式在粪便排泄;静脉注射主要以一部分原形和代谢产物G-Rc-Mb和G-Rc-Mc形式经尿液排泄[66]。
静脉或灌胃大鼠G-Rd,以原形或其代谢产物形式排泄[68]。给小鼠静脉注射G-Rd,小鼠给药后0~6、6~24、24~48、48~72h尿中累积排泄率分别为14.9%、60.8%、62.86%、63.36%;粪便中累积排泄率分别为7.20%、18.45%、18.72%、18.85%,主要经尿液排泄。大鼠静脉给药后0~12、12~24、24~48h尿中累积排泄率分别为30.5%、37.2%、39.5%;粪便中累积排泄率分别为15.6%、31.7%、36.6%,尿液和粪便皆是主要排泄途径[53]。总的来看,给药后24h主要经尿液清除。
给大鼠按0.5mg·kg-1剂量静脉注射20(R)-G-Rg3,主要以原形形式经胆汁排泄;进入肠道的20(R)-G-Rg3在肠道菌群的作用下降解产生苷元原人参二醇[56]。给大鼠按1.5mg·kg-1剂量肌注20(S)-G-Rg3,其主要经尿液排泄,给药8h内,累积经尿液排出的20(S)-G-Rg3占给予总量的72.1%,经胆汁排泄的20(S)-G-Rg3占给予总量的10.1%,大部分20(S)-G-Rg3经尿液由肾脏排出体外,在体内基本无蓄积[58]。
20(R)-G-Rh2在Beagle犬和大鼠的系统清除率分别低至约2、20mL·min-1·kg-1,实验中测定在Beagle犬的系统清除率为(1±0.5)mL·min-1·kg-1,恰好是Beagle犬一般肝血流的7%,说明20(R)-G-Rh2的体清除低。大鼠灌胃20(R)-G-Rh2在排泄物中的原形化合物的回收率仅为给予剂量的1%,静脉注射原形化合物在胆汁中的回收率为30%,这与20(R)-G-Rh2代谢产物的生成密切相关。详细的研究表明,按0.1mg·kg-1剂量给大鼠静脉注射20(R)-G-Rh2,原形化合物在给药后0~1、0~3、0~5、0~7、0~9、0~12h在胆汁中的累积排泄率分别为(13.00±2.46)%、(20.36±2.35)%、(23.69±2.51)%、(25.42±2.52)%、(26.73±2.67)%和(27.72±2.74)%。按3mg·kg-1剂量给大鼠灌胃20(R)-G-Rh2,原形化合物在药后0~2、0~4、0~6、0~8、0~10、0~12h在胆汁中的累积排泄率分别为(0.05±0.04)%、(0.23±0.27)%、(0.33±0.25)%、(0.45±0.18)%、(0.49±0.24)%、(0.52±0.31)%;原形化合物在药后0~24、0~48、0~72h在粪便中的累积排泄率分别为(0.55±0.34)%、(0.65±0.35)%、(0.80±0.31)%[46]。
给大鼠无论是静脉注射还是灌胃给予G-Re,G-Re能以原形及其代谢产物形式经尿液排泄[73]。给ICR小鼠按1mg·kg-1剂量静脉给予G-Re,有较短的半衰期,雄性为(0.2±0.03)h,雌性为(0.5±0.08)h,迅速从体内清除[47]。
志愿受试者口服200mg G-Re片[75],在尿液中可检测到原形化合物及其代谢产物G-Rg1、G-F1、G-Rh1和PPT,说明它们皆可经尿液排泄。
给大鼠静脉注射G-Rg1,原形化合物及其代谢产物G-Rh1和G-F1的血中清除半衰期分别为1.82、5.87和6.87h;灌胃情况下,原形化合物及其代谢产物G-Rh1、G-F1和PPT的血中清除半衰期分别为2.25、6.73、5.44和5.06h[60]。
给大鼠静脉注射G-Rg1,给药后0~2h、2~4h和4~8h,原形化合物G-Rg1在胆汁中的排泄率分别为给予剂量的(36.21±8.60)%、(18.80±7.37)%和(5.76±1.99)%,总排泄率为(60.77±6.14)%;给药后0~2h、2~4h、4~8h和8~12h,代谢产物G-Rh1在胆汁中的排泄率分别为给予剂量的(0.06±0.08)%、(0.55±0.46)%、(1.06±0.32)%和(0.57±0.28)%,总排泄率为(2.24±0.62)%;给药后2~4h、4~8h和8~12h,代谢产物G-F1在胆汁中的排泄率分别为给予剂量的(0.43±0.24)%、(0.27±0.15)%和(1.52±0.32)%,总排泄率为(1.52±0.32)%;给药后4~8h和8~12h,代谢产物PPT在胆汁中的排泄率分别为给予剂量的(0.16±0.12)%和(0.05±0.03)%,总排泄率为(0.21±0.13)%[60]。
给大鼠静脉注射G-Rg1,给药后0~4h、4~8h、8~12h,原形化合物G-Rg1在尿液中的排泄率分别为给予剂量的(15.74±6.08)%、(9.82±4.17)%和(2.39±0.53)%,总排泄率为(27.95±8.17)%;在粪便中的排泄率分别为给予剂量的(15.92±1.73)%、(1.31±0.21)%和(0.41±0.17)%,总排泄率为(17.64±2.42)%。0~4h、4~8h、8~12h和12~24h,代谢产物G-Rh1在尿液中的排泄率分别为给予剂量的(14.88±1.95)%、(3.17±1.01)%、(1.05±0.34)%、(0.16±0.06)%,总排泄率为(19.26±2.98)%;在粪便中的排泄率分别为给予剂量的(18.63±2.97)%、(5.47±1.48)%、(2.02±0.61)%、(0.11±0.36)%,总排泄率为(16.23±5.06)%;代谢产物G-F1在尿液中的排泄率分别为给予剂量的(15.76±2.02)%、(6.08±1.53)%、(0.63±0.15)%和(0.25±0.07)%,总排泄率为(12.72±3.75)%;在粪便中的排泄率分别为给予剂量的(17.74±1.69)%、(4.12±2.08)%、(0.96±0.24)%和(0.04±0.02)%,总排泄率为(12.86±2.79)%。在0~4h、4~8h和8~12h,代谢产物PPT在尿液中的排泄率分别为给予剂量的(10.18±0.11)%、(1.17±0.19)%和(0.03±0.01)%,总排泄率为(11.38±0.65)%;在0~4h、4~8h、8~12h和12~24h,代谢产物PPT在粪便中的排泄率分别为给予剂量的(14.19±2.02)%、(4.68±1.57)%、(1.72±0.36)%和(0.14±0.02)%,总排泄率为(10.73±4.59)%[60]。
总之,给大鼠静脉注射G-Rg1,原形化合物G-Rg1及其代谢产物在大鼠尿液和粪便中的总排泄率分别为51.31%和47.46%,主要排泄途径为胆汁[60]。
给大鼠静脉注射G-Rg2后,5.5h内胆汁中原形G-Rg2累积排泄率为给予剂量的27.2%,24h内粪便中原形G-Rg2累积排泄率为给予剂量的22.6%;尿液中未检出G-Rg2。由此可见,静脉给予大鼠G-Rg2,原形药物主要通过胆汁和粪便途径排出体外[85]。
按100mg·kg-1剂量给大鼠灌胃NPD,给药后96h,累积粪便排泄率为给予剂量的(64.56±20.32)%,尿液排泄率仅为(0.0233±0.0356)%,说明灌胃NPD的主要排泄途径为粪便[61]。
6 结语
尽管人参的三萜主要化学成分类型局限在达玛烷型和齐墩果酸型,但它们具有多样性的化学结构,具有不同的PK特性,决定了人参多样性的生物学活性和药理学作用。为了阐明人参的生物学活性、药理学作用、作用机制和临床应用,许多科学工作者专注于人参单体皂苷的研究,取得了可喜成绩。一般地说,脱糖基人参皂苷的疏水性和穿过细胞壁的能力随着脱糖基作用而增强,最终表现为生物学活性和药理学作用的增强,或发生作用性质的改变,人参皂苷结构的肠内细菌生物转化,在某种程度上活化了其作用。因此,天然的人参皂苷似乎是一种前药。实际上,口服给予的人参皂苷在肠道内的水解是不完全的,如将分子结构中具有3个糖基的G-Re口服给予健康志愿受试者,在尿液中可检测到结合三个糖基的原形化合物G-Re、结合2个糖基的G-Rg1、结合1个糖基的G-Rh1和G-F1及它们的苷元PPT[75]。在大鼠试验中亦得到了类似的结果[73]。给予健康志愿受试者口服人参标准提取物GinsanaG115胶囊(主要成分为G-Re和Rg1,还含有G-Rb1、Rb2、Rc、Rd、Rf等),在给药后0~3h累积尿液中可检测到G-Rb2、Rc、Rd、Re和Rg1;3~6h累积尿液中可检测到G-Rh1;6~12h累积尿液中可检测到G-Rb1和G-CK;12~24h累积尿液中可检测到G-Rh1/G-F1和G-CK[86]。从这个人体的试验结果,告诫我们要正确的理解体外人肠内细菌转化人参皂苷的实验结果,一些人参皂苷可以进入体循环发挥其生物学作用。尽管人参皂苷的组织分布研究较少,但某些人参皂苷可以分布到达心和脑组织,此可能是人参滋养心肌和益智的基础。此外,如果把原形人参皂苷的吸收和其代谢产物的吸收进行加和计算,将会改变人们对人参皂苷生物利用度差的认识。PPD和PPT型皂苷和/或皂苷元存在20R和20S差向异构体,同一对差向异构体有时具有差异性的PK性质,表现为生物学活性和药理学作用的差异性。过去几年人参单体皂苷结构上的变化导致其生物转化和/或代谢及PK特性变化的研究相对比较多,由于人参的应用多为混合人参皂苷的应用,今后应对人参皂苷相互作用及与其他中药成分相互作用导致其PK性质的变化展开研究,其成果可扩展到循证人参配伍复方作用机制上的应用。人参PK和生物学活性、药理学作用的研究进展亦将极大地激发人们对人参有用生物转化和/或代谢产物的合成[87-88]。
[1] 杨鑫宝,杨秀伟,刘建勋.人参中皂苷类化学成分的研究[J].中国现代中药,2013,13(5):349-358.
[2] 李珂珂,杨秀伟.人参茎叶化学成分的研究进展[J].中国现代中药,2012,14(1):47-50.
[3] 杨秀伟.人参中三萜类化学成分的研究[J].中国现代中药,2016,18(1):7-15.
[4] 杨秀伟,富力.人参中三萜类化学成分的生物学活性和药理学作用[J].中国现代中药,2016,18(1):36-55.
[5] Akao T,Kida H,Kanaoka M,et al.Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1fromPanaxginseng[J].J Pharm Pharmacol,1998,50(10):1155-1160.
[6] Xu R J,Peng Y,Wang M Y,et al.Effects of broad-spectrum antibiotics on the metabolism and pharmacokinetics of ginsenoside Rb1:A study on rats’ gut microflora influenced by lincomycin[J].J Ethnopharmacol,2014,158(Part A):338-344.
[7] Kanaoka M,Akao T,Kobashi K.Metabolism of ginseng saponins,ginsenosides,by human intestinal flora[J].J Tradit Med,1994,11(3):241-245.
[8] Shen H,Leung W I,Ruan J Q,et al.Biotransformation of ginsenoside Rb1viathe gypenoside pathway by human gut bacteria[J].Chin Med(London,United Kingdom),2013,8:22/1-22/11.
[9] Bae E A,Park S Y,Kim D H.Constitutiveβ-glucosidases hydrolyzing ginsenoside Rb1and Rb2from human intestinal bacteria[J].Biol Pharm Bull,2000,23(12):1481-1485.
[10] Karikura M,Miyase T,Tanizawa H,et al.Studies on absorption,distribution,excretion and metabolism of ginseng saponins.VII.Comparison of the decomposition modes of ginsenoside-Rb1and-Rb2in the digestive tract of rats[J].Chem Pharm Bull,1991,39(9):2357-2361.
[11] Hasegawa H,Sung J H,Benno Y.Role of human intestinalPrevotellaorisin hydrolyzing ginseng saponins[J].Planta Med,1997,63(5):436-440.
[12] Sung J H,Hasegawa H,Ha J Y,et al.Metabolism of ginseng saponins by human intestinal bacteria(part II)[J].Saengyak Hakhoechi,1997,28(1):35-41.
[13] Hasegawa H,Sung J H,Matsumiya S,et al.Main ginseng saponin metabolites formed by intestinal bacteria[J].Planta Med,1996,62(5):453-457.
[14] Bae E A,Choo M K,Park E K,et al.Metabolism of ginsenoside Rc by human intestinal bacteria and its related antiallergic activity[J].Biol Pharm Bull,2002,25(6):743-747.
[15] Shin J E,Park E K,Kim E J,et al.Cytotoxicity of compound K(IH-901)and ginsenoside Rh2,main biotransformants of ginseng saponins bybifidobacteria,against some tumor cells[J].J Ginseng Res,2003,27(3):129-134.
[16] Bae E A,Han M J,Choo M K,et al.Metabolism of20(S)-and20(R)-ginsenoside Rg3by human intestinal bacteria and its relation toinvitrobiological activities[J].Biol Pharm Bull,2002,25(1):58-63.
[17] Bae E A,Han M J,Kim E J,et al.Transformation of ginseng saponins to ginsenoside Rh2by acids and human intestinal bacteria and biological activities of their transformants[J].Arch Pharm Res,2004,27(1):61-67.
[18] Ko S R,Suzuki Y,Suzuki K,et al.Marked production of ginsenosides Rd,F2,Rg3,and compound K by enzymatic method[J].Chem Pharm Bull,2007,55(10):1522-1527.
[19] Park E K,Choo M K,Kim E J,et al.Antiallergic activity of ginsenoside Rh2[J].Biol Pharm Bull,2003,26(11):1581-1584.
[20] Bae E A,Shin J E,Kim D H:Metabolism of ginsenoside Re by human intestinal microflora and its estrogenic effect[J].Biol Pharm Bull2005,28(10):1903-1908.
[21] Chi H,Ji G E:Transformation of ginsenosides Rb1and Re fromPanaxginsengby food microorganisms.Biotechnol Lett,2005,27(11):765-771.
[22] Odani T,Tanizawa H,Takino Y.Studies on the absorption,distribution,excretion and metabolism of ginseng saponins.IV.Decomposition of ginsenoside-Rg1and-Rb1in the digestive tract of rats[J].Chem Pharm Bull,1983,31(10):3691-3697.
[23] 陈英杰,荻原幸夫.20(S)-人参皂苷-Rg2的代谢产物研究[J].沈阳药学院学报,1987,4(3):202.
[24] Wang H Y,Hua H Y,Liu X Y,et al.Invitrobiotransformation of red ginseng extract by human intestinal microflora:Metabolites identification and metabolic profile elucidation using LC-Q-TOF/MS[J].J Pharm Biomed Anal,2014,98:296-306.
[25] Liu H F,Yang J L,Du F F,et al.Absorption and disposition of ginsenosides after oral administration ofPanaxnotoginsengextract to rats[J].Drug Metabo Dispos,2009,37(12):2290-2298.
[26] Han M,Sha X Y,Wu Y J,et al.Oral absorption of ginsenoside Rb1usinginvitroandinvivomodels[J].Planta Med,2006,72(5):398-404.
[27] 韩旻,韩丽妹,王青松,等.三七皂苷的口服吸收机制[J].药学学报,2006,41(6):498-505.
[28] Liu C,Hu M Y,Guo H F,et al.Combined contribution of increased intestinal permeability and inhibited deglycosylation of ginsenoside Rb1in the intestinal tract to the enhancement of ginsenoside Rb1exposure in diabetic rats after oral administration[J].Drug metabolism and disposition:the biological fate of chemicals,2015,43(11):1702-1710.
[29] 赵洁,杨彩华,胡明,等.人参皂苷Rb2透过Caco-2单细胞层的吸收特征研究[J].南方医科大学学报,2009,29(12):2387-2390.
[30] 赵洁,杨彩华,胡明,等.人参皂苷Rb3透过Caco-2单细胞层模型上的吸收特征研究[J].中国药房,2010,21(3):196-198.
[31] 谢海棠,王广基,赵小辰,等.Caco-2细胞对人参皂苷Rg3的摄取及代谢研究[J].中国临床药理学与治疗学,2004,9(3):257-260.
[32] Xie H T,Wang G J,Chen M,et al.Uptake and metabolism of ginsenoside Rh2and its aglycon protopanaxadiol by Caco-2cells[J].Biol Pharm Bull,2005,28(2):383-386.
[33] Gu Y,Wang G J,Wu X L,et al.Intestinal absorption mechanisms of ginsenoside Rh2:stereoselectivity and involvement of ABC transporters[J].Xenobiotica,2010,40(9):602-612.
[34] 顾轶,王广基,张经纬,等.20(R)-人参皂苷Rh2的大鼠肠吸收动力学[J].中国临床药理学与治疗学,2009,14(4):368-373.
[35] Paek I B,Moon Y,Kim J,et al.Pharmacokinetics of a ginseng saponin metabolite compound K in rats[J].Biopharm Drug Dispos,2006,27(1):39-45.
[36] Li N,Wang D D.Ge G B,et al.Ginsenoside metabolites inhibitP-glycoproteininvitroandinsituusing three absorption models[J].Planta Med,2014,80(4):290-296.
[37] Yang Z,Wang J R,Niu T,et al.Inhibition ofP-glycoprotein leads to improved oral bioavailability of compound K,an anticancer metabolite of red ginseng extract produced by gut microflora[J].Drug Metabo Dispos,2012,40(8):1538-1544.
[38] Liang Y,Zhou Y Y,Zhang J W,et al.Pharmacokinetic compatibility of ginsenosides andSchisandralignans in Shengmai-san:from the perspective ofP-glycoprotein[J].PLoS One,2014,9(6):e98717/1-e98717/12.
[39] 李昊,孙建国,谢海棠,等.大鼠肠管外翻模型对人参皂苷Rg1吸收机制的研究[J].中国临床药理学与治疗学,2004,9(5):510-513.
[40] Wakabayashi C,Hasegawa H,Murata J,et al.Invivoantimetastatic action of ginseng protopanaxadiol saponins is based on their intestinal bacterial metabolites after oral administration[J].Oncol Res,1997,9(8):411-417.
[41] Leea J,Leea E,Kim D,et al.Studies on absorption,distribution and metabolism of ginseng in humans after oral administration[J].J Ethnopharmacol,2009,122(1):143-148.
[42] Xu Q F,Fang X L,Chen D F.Pharmacokinetics and bioavailability of ginsenoside Rb1and Rg1fromPanaxnotoginsengin rats[J].J Ethnopharmacol,2003,84(2-3):187-192.
[43] Li X Y,Wang G J,Sun J G,et al.Pharmacokinetic and absolute bioavailability study of total panax notoginsenoside,a typical multiple constituent traditional chinese medicine(TCM)in rats[J].Biol Pharm Bull2007,30(5):847-851.
[44] Wang W,Wang G J,Xie H T,et al.Determination of ginsenoside Rd in dog plasma by liquid Chromatography-mass spectrometry after solid-phase extraction and its application in dog pharmacokinetics studies[J].J Chromatogr B,2007,852(1-2):8-14.
[45] Xie H T,Wang G J,Sun J G,High performance liquid chromatographic-mass spectrometric determination of ginsenoside Rg3and its metabolites in rat plasma using solid-phase extraction for pharmacokinetic studies[J].J Chromatogr B,2005,818(2):167-173.
[46] Gu Y,Wang G J,Sun J G,et al.Pharmacokinetic characterization of ginsenoside Rh2,an anticancer nutrient from ginseng,in rats and dogs[J].Food Chem Toxicol,2009,47(9):2257-2268.
[47] Joo K M,Lee J H,Jeon H Y,et al.Pharmacokinetic study of ginsenoside Re with pure ginsenoside Re and ginseng berry extracts in mouse using ultra performance liquid chromatography/mass spectrometric method[J].J Pharm Biomed Anal2010,51(1):278-283.
[48] Sun J G,Wang G J,Xie H T,Simultaneous rapid quantification of ginsenoside Rg1and its secondary glycoside Rh1and aglycone protopanaxatriol in rat plasma by liquid chromatography-mass spectrometry after solid-phase extraction[J].J Pharm Biomed Anal,2005,38(1):126-132.
[49] Xu M J,Wang G J,Xie H T,et al.Determination of ginsenoside Rg2in rat plasma by high-performance liquid chromatography-mass spectrometry after solid-phase extraction[J].Analytical Letters,2006,39(1):113-126.
[50] Lai L,Hao H P,Liu Y T,et al.Characterization of pharmacokinetic profiles and metabolic pathways of20(S)-ginsenoside Rh1invivoandinvitro[J].Planta Med,2009,75(8):797-802.
[51] Zhang X R,Zhang D,Xu J H,et al.Determination of25-OH-PPD in rat plasma by high-performance liquid chromatography-mass spectrometry and its application in rat pharmacokinetic studies[J].J Chromatogr B,2007,858(1-2):65-70.
[52] Qi L W,Wang C Z,Yuan C S:Isolation and analysis if ginseng:advances and challenges[J].Nat Prod Rep,2011,28(3):467-495.
[53] Sun D,Wang B,Shi M,et al.Pharmacokinetic,tissue distribution and excretion of ginsenoside-Rd in rodents[J].Phytomedicine,2012,19(3-4):369-373.
[54] 杨秀伟.中药成分的吸收、分布、代谢、排泄、毒性与药效(ADME/Tox./Act):上册[M].北京:中国医药科技出版社,2006:978-1052.
[55] Bae S H,Park J B,Zheng Y F,et al.Pharmacokinetics and tissue distribution of ginsenoside Rh2and Rg3epimers after oral administration of BST204,a purified ginseng dry extract,in rats[J].Xenobiotica,2014,44(12):1099-1107.
[56] 李柯.薯蓣皂苷和人参皂苷Rg3动物体内药物动力学研究[D].沈阳:沈阳药科大学,2005.
[57] 刘继华,卢丹,刘金平,等.20(S)-人参皂苷Rg3眼膏在兔眼组织分布及其药代动力学[J].药学学报,2005,40(3):258-261.
[58] 刘继华,卢丹,刘金平,等.大鼠肌内注射20(S)-人参皂苷Rg3的药动学研究[J].中国药学杂志,2007,42(14):1087-1090.
[59] 张均田.人参冠百草—人参化学、生物活性和药代动力学研究进展[M].北京:化学工业出版社,2008,72-78.
[60] Feng L,Wang L,Hu C J,et al.Pharmacokinetics,tissue distribution,metabolism,and excretion of ginsenoside Rg1in rats[J].Arch Pharm Res,2010,33(12):1975-1984.
[61] 刘梅,王莉,胡凯莉,等.人参皂苷Rg1经PEG修饰前后的组织分布研究[J].中国中药杂志,2012,37(12):1747-1750.
[62] Geng C,Yin J Y,Yu X H,et al.Tissue distribution and excretion study of neopanaxadiol in rats by ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry[J].Biomed Chromatogr,2015,29(3):333-340.
[63] Hao M,Wang W,Zhao Y Q,et al.Pharmacokinetics and tissue distribution of25-hydroxyprotopanaxadiol,an anti-cancer compound isolated fromPanaxginseng,in athymic mice bearing xenografts of human pancreatic tumors[J].Eur J Drug Metabo Pharmacokinet,2011,35(3-4):109-113.
[64] Chen GT,Yang M,Song Y,et al.Comparative analysis on microbial and rat metabolism of ginsenoside Rb1by high-performance liquid chromatography coupled with tandem mass spectrometry[J].Biomed Chromatogr,2008,22(7):779-785.
[65] 林力,刘建勋,张颖,等.LC-MS/MS法测定大鼠口服人参皂苷Rg1,Re,Rb1和Rd 的药动学[J].中国药学杂志,2009,44(5):373-377.
[66] Sun J H,Wu W,Guo Y Y,et al.Pharmacokinetic study of ginsenoside Rc and simultaneous determination of its metabolites in rats using RRLC-Q-TOF-MS[J].J Pharm Biomed Anal,2014,88:16-21.
[67] 杨柳,许舜军,曾星,等.大鼠尿中人参皂苷Rd及其代谢物的LC-MS研究[J].药学学报,2006,41(8):742-746.
[68] Yang L,Deng Y H,Xu S J,et al.Invivopharmacokinetic and metabolism studies of ginsenoside Rd[J].J Chromatogr B,2007,854(1-2):77-84.
[69] Qian TX,Cai ZW,Wong RNS,et al.Invivorat metabolism and pharmacokinetic studies of ginsenoside Rg3[J].J Chromatogr B,2005,816(1-2):223-232.
[70] Li K,Chen X Y,Xu J H,Li X,et al.Liquid chromatography/tandem mass spectrometry for pharmacokinetic studies of20(R)-ginsenoside Rg3in dog[J].Rapid Commun Mass Spectrom,2005,19(6):813-817.
[71] Wang H L,Zou H F,Kong L,et al.Determination of ginsenoside Rg3in plasma by solid-phase extraction and high-performance liquid chromatography for pharmacokinetic study[J].J Chromatogr B,1999,731(2):403-409.
[72] 庞焕,汪海林,富力,等.20(R)-人参皂苷Rg3人体药代动力学研究[J].药学学报,2001,36(3):170-173.
[73] Yang L,Xu S J,Liu C J,et al:Invivometabolism study of ginsenoside Re in rat using high-performance liquid chromatography coupled with tandem mass spectrometry[J].Anal Bioanal Chem2009,395(5):1441-1453.
[74] 彭缨,王淑君,潘卫三,等.人参皂苷Re大鼠体内药物动力学研究[J].沈阳药科大学学报,2006,23(4):197-200.
[75] Liu L,Huang J Q,Hu X,et al.Simultaneous determination of ginsenoside(G-Re,G-Rg1,G-Rg2,G-F1,G-Rh1)and protopanaxatriol in human plasma and urine by LC-MS/MS and its application in a pharmacokinetics study of G-Re in volunteers[J].J Chromatogr B,2011,879(22):2011-2017.
[76] Yang L,Xu S J,Wu Z F,et al.Determination of ginsenoside-Rg1in human plasma and its application to pharmacokinetic studies following intravenous administration of ‘Shenmai’ injection[J].Phytother Res,2009,23(1):65-71.
[77] 杨秀伟,桂方晋,田建明,等.人参皂苷-Rg2在大鼠体内的药代动力学[J].中国药理学通报,2009,25(7):967-970.
[78] Gui FJ,Yang XW,Li LY,et al.Simultaneous enantiomer determination of20(R)-and20(S)-ginsenoside-Rg2in rat plasma after intravenous administration using HPLC method[J].J Chromatogr B,2007,850(1-2):1-6.
[79] Kong L T,Wang Q,Xiao B X,et al.Different pharmacokinetics of the two structurally similar dammarane sapogenins,protopanaxatriol and protopanaxadiol,in rats[J].Fitoterapia,2013,86:48-53.
[80] Zhao J,Su C,Yang C P,et al.Determination of ginsenosides Rb1,Rb2,and Rb3in rat plasma by a rapid and sensitive liquid chromatography tandem mass spectrometry method:Application in a pharmacokinetic study[J].J Pharm Biomed Anal,2012,64-65:94-97.
[81] Chen W,Dang Y J,Zhu C Y.Simultaneous determination of three major bioactive saponins ofPanaxnotoginsengusing liquid chromatography-tandem mass spectrometry and a pharmacokinetic study[J].Chin Med,2010,5:12-17.
[82] Song M,Zhang S Y,Xu X Y,et al.Simultaneous determination of threePanaxnotoginsengsaponins at sub-nanograms by LC-MS/MS in dog plasma for pharmacokinetics of compound Danshen tablets[J].J Chromatogr B,2010,878(32):3331-3337.
[83] Deng G F,Wang D L,Meng M X,et al.Simultaneous determination of notoginsenoside R1,ginsenoside Rg1,Re,Rb1and icariin in rat plasma by ultra-performance liquid chromatography-tandem mass spectrometry[J].J Chromatogr B,2009,877(22):2113-2122.
[84] Li Z G,Zhang R,Wang X P,et al.Simultaneous determination of seven ginsenosides in rat plasma by high-performance liquid chromatography coupled to time-of-flight mass spectrometry:application to pharmacokinetics of Shenfu injection[J].Biomed Chromatogr,2015,29(2):167-175.
[85] 杨秀伟,桂方晋,宋燕,等.人参皂苷-Rg2的排泄实验研究[J].中国中药杂志,2009,34(10):1281-1284.
[86] Tawab M A,Bahr U,Karas M,et al.Degradation of ginsenosides in humans after oral administration[J].Drug Metab.Dispos.2003,31(8):1065-1071.
[87] 吕静,李珂珂,李争宁,等.人参皂苷化合物K乙酰化衍生物的合成及分析[J].中国现代中药,2015,17(5):431-439.
[88] Li W F,Chen L R,Gong X J,et al.Synthesis of esters of ginsenoside metabolite M1and their cytotoxicity on MGC80-3cells[J].Molecules,2013,18(4):3689-3702.
PharmacokineticStudiesofChemicalConstituentsofGinseng
YANGXiuwei*
(StateKeyLaboratoryofNaturalandBiomimeticDrugs,DepartmentofNaturalMedicines,SchoolofPharmaceuticalSciences,PekingUniversity,Beijing100191,China)
The wide range of therapeutic and health care potential of ginseng has been studied extensively,and ginsenosides,the principal active ingredients of ginseng,are shown to be involved in modulating multiple physiological activities.Biological and environmental factors may affect the efficacy of ginsenosides.Evidence from pharmacokinetic and metabolic studies of ginsenosides demonstrated that (1) the absorption of many ginsenosides from gastrointestinal tract is different along with their different degrees of glycosylation in the molecular structure;(2) the poor membrane permeability of polyglycosylated prototypic ginsenoside predicted from the human Caco-2cell monolayer model restricted their oral absorption badly;(3) ginsenoside may be metabolized mainly to their prosaponins and/or aglycones by intestinal microflora before absorption into circulatory system;(4) the intestinal microflora-dependent biotransformation of some ginsenosides in the gastrointestinal tract could reflect a pathway of bioactivation resulting in the formation of bioactive intermediates and/or end-products,suggesting that ginsenoside is likely to be a prodrug;and (5) intact and some deglycosylated products are cleared from the body.This review provides an overview of the recent advances in biotransformation and/or metabolism as well as pharmacokinetics of ginsenosides,which might greatly contribute to supply the scientific basis for Evidence-Based Medicine in clarifying the effective substance basis of ginseng.
Panaxginseng;ginsenoside;pharmacokinetic
10.13313/j.issn.1673-4890.2016.1.004
2015-10-30)
国家自然科学基金重点项目(81530097);“十二五”国家科技支撑专项(2011BAI03B01;2011BAI07B08)
*
杨秀伟,教授,博士生导师,研究方向:天然产物化学与药物代谢;E-mail:xwyang@bjmu.edu.cn
