不完全型雄激素不敏感综合征产前诊断研究*
2016-04-04吴维青文华轩谢建生
吴维青 文华轩 袁 晖 谢建生**
1. 南方医科大学附属深圳妇幼保健院中心实验室(深圳 518032);
2. 南方医科大学附属深圳妇幼保健院超声科;3. 南方医科大学附属深圳妇幼保健院产科
·论 著·
不完全型雄激素不敏感综合征产前诊断研究*
吴维青1文华轩2袁 晖3谢建生1**
1. 南方医科大学附属深圳妇幼保健院中心实验室(深圳 518032);
2. 南方医科大学附属深圳妇幼保健院超声科;3. 南方医科大学附属深圳妇幼保健院产科
目的对不完全型雄激素不敏感综合征(Partial Androgen Insensitivity Syndrome, PAIS)一家系进行病例报道及遗传分析,并对高危胎儿进行产前诊断。方法分析患者雄激素受体(AR)基因序列及第一外显子内的CAG重复,结合核型及产前超声判断胎儿是否罹患PAIS。结果2名患者AR基因编码区及侧翼序列未见异常,男性胎儿获得了与疾病相关的CAG重复次数,超声检查提示其外生殖器发育异常。结论本研究对一个PAIS家系进行了遗传分析,虽未能明确AR基因突变,但连锁分析及产前超声均提示男性胎儿罹患PAIS。本研究可为同样病例的遗传分析、产前诊断和遗传咨询提供借鉴资料。
不完全型雄激素不敏感综合征;雄激素受体基因;产前诊断;连锁(遗传学)
Key woordspartial Androgen Insensitivity Syndrome(PAIS);androgen receptor gene;prenatal diagnosis;Linkage (Genetics)
雄激素不敏感综合征(ardrogen insenstivity syndromes, AIS)是由位于Xq11.12的雄激素受体基因(androgen receptor,AR)突变所致的X连锁隐性遗传病,根据表型轻重可分为完全型雄激素敏感综合征(complete androgen insensitivity syndrome, CAIS)和不完全型雄激素敏感综合征(partial androgeninsensitivity syndrome, PAIS, MIM #312300)。PAIS患者为男性,但具有程度不等的外生殖器发育异常(阴茎短小、尿道下裂)、生育功能低下、乳房发育等女性第二性征。PAIS发病率低于CAIS,但具体的发病频率尚不清楚[1]。
据统计,已报道的引起PAIS的AR基因(CAG)突变已超过100多种[2],主要位于编码AR蛋白非关键功能区域内。同一种突变在不同家系导致的临床表现轻重程度可明显不同,携带同一突变的同一家系成员临床表现也不尽相同。鉴于经典基因突变检测一般仅局限于外显子及侧翼序列等多种原因,并非所有AIS疑似患者均能检测到AR基因突变[3]。AR基因第一外显子内存在一个CAG三核苷酸重复,其重复次数的增加与脊髓延髓肌肉萎缩症、乳腺癌、前列腺癌、子宫及内膜癌、男性不育等多种疾病相关[4,5]。CAG重复次数范围在人群中呈明显的多态性,且具有种族差异,中国人此位点亦呈明显的多态性[6],有研究表明中国汉族人群CAG重复具有高度的遗传多态性(杂合度为0.833,多态信息量为0.88)[7],属于较好的连锁分析标志。
国内已有一些关于CAIS患者AR基因突变的报告[8,9],但对PAIS,却只有少数病例报告[10-12]及尿道下裂患者AR基因突变研究[13]。本研究对一个典型的PAIS家系的2名患者进行AR基因突变检测,综合利用连锁分析、遗传学性别鉴定及产前超声检查明确胎儿是否罹患PAIS,为同类病例的遗传学诊断及遗传咨询提供借鉴资料。
资料与方法
一、研究资料
一个PAIS家系(见图1),共有5名患者,呈现典型X连锁隐性遗传特征。先证者(Ⅳ7),男,7岁,出生时即发现阴茎短小、尿道下裂,于2岁时行尿道成形术,染色体核型为46, XY。先证者舅舅(Ⅲ5),男,34岁,出生时也有阴茎短小、尿道下裂,行阴茎成形矫正手术后可勃起;精液分析提示少精子、死精子;未能生育子女;多次因乳房发育行乳腺组织切除手术;其性激素6项检查结果显示睾酮(T)、雌二醇(E2)及促黄体生成素(LH)偏高,具体数值为:T 8.45μg/L(1.75~7.81)、孕酮(P)1.16μg/L(0.14~2.06)、E265pg/mL(20~47)、泌乳素(PRL)5.06μg/L(2.64~13.13),LH 13.90IU/L(1.24~8.6)、促卵泡生成素(FSH)12.43IU/L(1.3~19.3);染色体核型为46, XY。家系中其他患者均有类似情况,且均未能生育子女。家系遗传方式、患者的临床表现及实验室检查符合PAIS的诊断。先证者母亲(Ⅲ6),31岁,孕11周时来我院就诊,要求进行基因诊断和产前诊断,签署知情同意书,于孕17周行羊水穿刺术。
二、方法
1.DNA提取:外周血及羊水标本,以QIGEN试剂盒提取基因组DNA。
2.AR基因突变检测:根据AR基因序列,应用primer 5 软件自行设计引物,对2名患者AR基因8个外显子及测序序列进行分析,计数外显子1内CAG重复次数。
3.CAG重复连锁分析:根据AR基因exon1序列,参考文献[14]设计包含重复序列的一对引物,正向引物以荧光标记,产物于CEQ8000遗传分析仪上进行片段分析。检测患者、胎儿及家系中其他相关个体共7例(Ⅱ2、Ⅲ4、Ⅲ5、Ⅲ6、Ⅳ6、Ⅳ7、Ⅳ8)片段长度,判断重复次数在家系中是否具有多态性以及与疾病的关联情况。
4.G染色体核型分析:羊水细胞贴壁培养,之后常规G显带核型分析,判断胎儿遗性别及染色体是否正常。
5.胎儿超声检查:高危胎儿孕24周左右,以西门子512彩色超声成像仪进行胎儿三维超声检查,尤其关注胎儿内外生殖器发育情况。
结 果
一、AR基因突变检测结果
2名患者(Ⅲ5、Ⅳ7)AR基因各外显子及侧翼序列均未见碱基改变。
二、AR基因exon1片段分析结果
片段分析及测序结果提示2名患者(Ⅲ5、Ⅳ7)CAG重复均为21次(305bp)。Ⅱ2、Ⅲ4、 Ⅲ6、Ⅳ6、Ⅳ8片段长度为305/317、305/308、305/308、308、305bp。可以看出21次重复自Ⅱ2传递给Ⅲ4、Ⅲ5、Ⅲ6,男性个体Ⅲ5发病,而女性个体Ⅲ4、Ⅲ6携带,Ⅲ4未将其传递给后代Ⅳ6,而Ⅲ6传给儿子Ⅳ7及胎儿Ⅳ8。所有获得了21次重复的男性个体均有生殖器异常,表明其与疾病连锁共分离,见图1。
三、 G染色体核型分析结果
G显带核型分析结果显示胎儿为正常男性核型46,XY。
四、 胎儿24周超声检查结果
孕24周+6d,男性胎儿,阴茎偏小,形态异常,阴茎阴囊部分扭转伴随尿道下裂,见图2。
五、妊娠结局
孕妇知悉以上结果选择放弃胎儿,引产胎儿证实外生殖器确有尿道下裂等畸形。
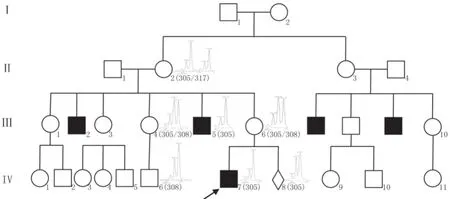
图1 PAIS家系图谱及连锁分析结果

图2 孕24周胎儿生殖器超声图像
讨 论
PAIS患者的表现轻重不同,临床诊断上相对困难。PAIS患者基础状态的T和LH较正常男性明显增高,因T可以转化为雌激素,故而多数患者E2水平也同时增高[15]。根据临床表现、典型的激素水平以及X连锁隐性遗传家族史,即可诊断为PAIS[15,16]。本研究家系呈现明显的X连锁隐性遗传特性,所有患者生殖器均发育不良,而且成年患者均有乳腺发育及不育,一例成年患者性激素T、LH、E2增高情况也符合PAIS特征,因此PAIS的诊断明确。
对此家系2名患者的AR基因进行了较为仔细的测序分析,遗憾的是未能找到致病基因突变。而根据相关研究,一半以上的PAIS患者不能明确基因异常位置[2],其原因可能是突变存在于AR基因的非编码区、内含子或者基因的调控区。随着现代测序技术的进步,后续研究可以对AR基因进行扩大范围检测,有望明确患者的基因变异。
PAIS患者社会性别为男性,症状较社会性别为女性的CAIS轻,但具有男性化不足、女性第二性征和生育功能缺陷等,给患者本人和家属带来更大、更长期的生理及心理负担,即使手术可以部分矫正异常,但也无法恢复到正常男性的生理功能[17]。当一个家庭出现PAIS患者时,明确突变类型并进行产前诊断是避免患儿出生和降低再发风险的唯一途径。
孕中期超声可以发现多种胎儿生殖器的异常[18],但生殖器畸形病因繁多,需进行鉴别诊断。有研究者认为,对于单纯尿道下裂胎儿来说,首先要考虑的就是部分型雄激素不敏感综合征(PAIS)存在的可能[19]。此高危胎儿核型分析明确为男性,超声发现其阴茎短小、阴茎阴囊部分扭转及尿道下裂,再结合家系遗传方式及两名患者的临床表现和实验室检查,可以确定胎儿罹患PAIS。
因基因突变无法明确这就给产前诊断带来了极大的困难,在这种情况下,对家系成员进行连锁分析并结合临床资料推断胎儿是否罹病,虽然是退而求其次的办法,但是对家系避免再次出现PAIS同样具有非常重要的意义。AR基因第一外显子CAG重复在中国人群属于高度多态的位点,并且就在致病基因内部,与疾病高度连锁,因此可视作AIS较好的连锁分析标记。我们发现CAG的21次重复(305bp)在此家系与PAIS表型共分离,高危男性胎儿恰巧获得了此标记,提示遗传到了与疾病连锁的AR基因,超声及引产发现的生殖器畸形也证实胎儿的确罹患PAIS,从而证实连锁分析的可靠性。国际上,也有应用此CAG重复鉴定致病AR基因,从而进行产前诊断的研究报告[20]。综述分析,应用AR基因exon1的CAG重复进行连锁分析,有益并且切实可行。
本研究对一个PAIS家系进行产前诊断,虽然没有能够找到AR基因突变,但结合AR基因内部的CAG重复次数与疾病的连锁关系及产前超声影像胎儿生殖器畸形情况,明确胎儿罹患部分型雄激素不敏感综合征,为不能明确突变类型的CAIS及PAIS家系产前诊断提供一个较好先例,对AIS的遗传学诊断和优生咨询均具有一定的借鉴意义。
1 Mendoza N, Motos MA. Androgen insensitivity syndrome.Gynecol Endocrinol2013;29(1):1-5
2 Gottlieb B, Beitel LK, Nadarajah A,et al. The Androgen Receptor Gene Mutations Database: 2012 Update.Hum Mutat2012;33(5): 1317-1327
3 Lek N, Miles H, Bunch T,et al. Low frequency of androgen receptor gene mutations in 46 XY DSD, and fetal growth restriction.Arch Dis Child2014;99(4):358-361
4 Rajender S, Singh L, Thangaraj K. Phenotypic heterogeneity of mutations in androgen receptor gene.Asian J Androl2007;9(2):147-179
5 任维果, 周自寅, 边家盛, 等. 前列腺鳞状细胞癌1例报告并文献复习. 中国男科学杂志 2008;22(10): 48-50
6 王钢, 陈光椿, 王晓慧, 等. 中国男性雄激素受体基因(CAG)n重复多态性的初步研究. 中华医学遗传学杂志2001;18(6): 456-458
7 张钏, 郭孟境, 裴利国, 等. 宁夏回、汉族群体雄激素受体基因(CAG)n、(GGN)n重复多态性研究. 中华医学遗传学杂志 2013;30(3): 365-369
8 吴维青, 罗福薇, 耿茜等. 完全型雄激素不敏感综合征雄激素受体基因突变分析. 中华医学遗传学杂志 2009;26(6): 606-609
9 信艳萍, 吴庆华, 张毅等. 两个雄激素不敏感综合征家系中AR基因突变检测. 郑州大学学报·医学版 2015;50(2): 202-206
10 张建平, 王晓春, 金杭美. 睾丸女性化综合征17例临床分析. 苏州大学学报·医学版 2009;29(1): 176-177,179
11 李莉平, 徐建平. 男性假两性畸形—不完全性雄激素不敏感综合征1例. 暨南大学学报·自然科学与医学版2002;23(2): 121
12 龚飞凤, 訾聃, 周遵伦, 等. 不完全性雄激素不敏感综合征1例. 贵阳医学院学报 2008;33(5): 565-566
13 李强, 李森恺, 徐家杰等. 尿道下裂患者雄激素受体基因突变的研究. 中华整形外科杂志 2004;20(6): 421-424
14 李座祥, 唐文豪, 汪朝晖等. 中国特发性无精子症和少精子症患者雄激素受体基因CAG重复多态性研究. 中华男科学杂志 2005;11(5): 335-338
15 Melo KF, Mendonca BB, Billerbeck AE,et al. Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: fi ve novel mutations in the androgen receptor gene.J Clin Endocrinol Metab2003;88(7):3241-3250
16 Gottlieb B, Pinsky L, Beitel LK,et al. Androgen insensitivity.Am J Med Genet1999;89(4):210-217
17 Rybar R, Markova P, Veznik Z,et al. Sperm chromatin integrity in young men with no experiences of infertility and men from idiopathic infertility couples.Andrologia2009;41(3): 141-149
18 Pajkrt E, Petersen OB, Chitty LS. Fetal genital anomalies: an aid to diagnosis.Prenat Diagn2008;28(5):389-398
19 Chitty LS, Chatelain P, Wolffenbuttel KP,et al.Prenatal management of disorders of sex development.J Pediatr Urol2012;8(6):576-584
20 Fogu G, Bertini V, Dessole S,et al. Identification of a mutant allele of the androgen receptor gene in a family with androgen insensitivity syndrome: detection of carriers and prenatal diagnosis.Arch Gynecol Obstet2004;269(4):266-269
(2016-05-08收稿)
Genetic analysis and prenatal diagnosis in a family with partial androgen insensitivity syndrome*
Wu Weiqing1, Wen Huaxuan2, Yuan Hui3, Xie Jiansheng1**
1. Central Laboratory, Southern Medical University Affiliated Shenzhen Maternity & Child Healthcare Hospital, Shenzhen 518028, Guangdong, China;2. Department of Ultrasonography, Southern Medical University Affiliated Shenzhen Maternity & Child Healthcare Hospital, Shenzhen;3. Department of Obstetrics, Southern Medical University Affiliated Shenzhen Maternity & Child Healthcare Hospital
Xie Jiansheng, Email:jianshengxie2000@aliyun.com
Objective To detect AR gene mutation in 2 patients of a partial androgen insensitivity syndrome family and perform prenatal diagnosis for the high risk fetus.MethodsEight exons and at least 100bp fl anking intrinsic sequence of AR gene were screened by PCR and direct sequencing. CAG repeats in exon1 of AR gene was used as a STR marker in linkage analysis. The amniocentesis was performed for determination of genetic gender and linkage analysis. The Fetal genital anomalies were scanned by color Doppler ultrasound.ResultsNo mutation was found in two patients' AR gene. Linkage analysis indicated that the 21 times of CAG repeats was related to the phenotype in this family. The male fetus inherited the 21 times repeat of CAG, and antenatal sonographic diagnosis showed its external genitals was abnormal(micropenis, perineoscrotal hypospadia).ConclusionMale fetus was still diagnosed as PAIS based on the results of linkage analysis and sonographic diagnosis, although no specific mutations of AR gene were detected in PAIS patients.
10.3969/j.issn.1008-0848.2016.06.001
R714.55
资助:本课题受深圳市科技研发资金基础研究项目(JCYJ201504020904413001);国家自然科学基金项目(31471204)
**通讯作者,E-mail:jianshengxie2000@aliyun.com
