固相萃取—超高效液相色谱—串联质谱法同时检测地表水中的35种农药及降解产物
2015-08-13孙静等
孙静等


摘 要 采用固相萃取(SPE)超高效液相色谱串联质谱(UPLCMS/MS)技术,建立了同时测定环境水样中痕量浓度多极性的35种高关注农药及转化产物的方法。优化的前处理方法为:串联HLB和活性炭小柱用于富集水样,10 mL乙腈为HLB柱洗脱溶剂,10 mL 20 mmol/L乙酸铵甲醇作为活性炭柱的洗脱溶剂,将洗脱溶液混合、浓缩、溶剂转化和定容,定容溶剂为10 mmol/L乙酸铵的乙腈水的混合溶液(1∶1, V/V)。流动相分别为5 mmol/L甲酸铵甲酸缓冲溶液(pH 3.5)和乙腈进行UPLC色谱分离,质谱用多反应监测(MRM)模式进行分析测定。实验结果表明,目标物在0.5~200 μg/L含量范围内线性良好(R2≥0.9802),检出限范围为0.01~0.8 ng/L,回收率在60%~120%之间。本研究构建的SPEUPLCMS/MS方法灵敏可靠,可用于对地表水中ng/L级极性不同的多种农药及转化产物的测定。
关键词 ;农药; 转化产物; 固相萃取; 超高效液相色谱串联质谱法; 地表水
1 引 言
农药的大量使用造成各种环境基质中存在农药残留。农药对地表水形成污染的主要原因在于大部分残留在土壤或漂浮于大气中的农药通过雨水沉降和地表径流的方式进入地表水。农药在环境中通过水解、氧化、生物降解和光解作用形成转化产物[1]。转化产物具有比母体化合物更强的极性和水溶性,其在地表水高浓度检出和潜在的健康风险引起人们广泛关注[1~3]。部分农药及转化产物已被美国环保局(US EPA)列入饮用水污染物候选清单中(CCL3)中,如乙酰甲胺磷、甲胺磷、乐果、灭多威、异丙威、特丁磷砜、戊唑醇、草达灭、虫酰肼和乙草胺等。目前,国内针对地表水中的农药污染调查尚不系统和全面[4]。6种高毒有机磷品种(敌敌畏、甲基对硫磷、对硫磷、氧化乐果、甲胺磷、久效磷)的产量占国内有机磷杀虫剂产量的70%,占农药总产量的33%[5],但《国内地表水环境质量标准(GB38382002)》中仅规定了前3种有机磷农药(敌敌畏、对硫磷、甲基对硫磷)的标准限值,尚未对后3种强极性有机磷农药做出限值规定。研究表明,甲胺磷、氧化乐果等有机磷农药具有强极性特征,其辛醇水分配系数小于零,广泛存在于地表水中[6]。
农药种类繁多,各类农药的极性差别较大,因而分析方法也显著不同。针对环境水样中的有机磷农药或转化产物,分析方法主要为气相色谱法,检测器为氢火焰离子化检测器[7]或火焰光度检测器[6]。气相色谱串联质谱法(GCMS/MS)[8~10]在测定有机磷转化产物时也被使用,但在测定前需衍生化。液相色谱质谱法(LCMS)[11]或液相色谱串联质谱法(LCMS/MS)[12~14]可用于检测分析环境水样中多类别农药或转化产物。与GCMS相比,LCMS/MS方法在测定农药种类范围及检测灵敏度方面更具优势[15]。与固相微萃取法(SPME)[16]和液相微萃取法(LPME)[17]相比,固相萃取法(SPE)有较高的富集倍数,是目前富集水样中农药转化产物主要的前处理方法[18]。富集农药及转化产物的商业化和通用性的SPE小柱主要有C18柱[11,19],聚合柱[20,21]和石墨化炭黑柱[22]。聚合柱因为同时含有亲水基团和疏水基团,对多数农药有较好的富集效果[21,23],但是对于极性特强的有机磷农药(如乙酰甲胺磷、甲胺磷、氧化乐果)的萃取效率低于15%[24,25]。活性炭柱已被应用于富集水中丙烯酰胺[26]、二甲基亚硝胺[27]等强极性化合物。Hayama等[28]利用活性炭柱固相萃取亲水作用色谱分离测定环境水样中的6种强极性有机磷农药,回收率在76.4%~98.6%之间。然而,活性炭柱具有较强的吸附能力,水体中的天然有机物[29]、痕量有机污染物[29]和重金属离子[30]均被富集在萃取小柱上,大量干扰物的存在可能会影响强极性有机磷农药在UPLCMS/MS上的色谱分离和质谱分析。
针对以上问题,本研究优化聚合柱,串联活性炭柱,减少杂质的吸附,同时提高极性较弱的农药的富集效率,建立一种同时测定环境水样中浓度在ng/L水平的极性跨度大Symbolm@@ 0.85≤lgKow≤5.2)的35种农药及转化产物的SPEUPLCMS/MS方法。本实验选择了国内产量较高的农药,特别是强极性有机磷农药、列入US EPA CCL3中的部分农药及转化产物,作为目标化合物。采用HLB柱和活性炭柱串联,可用于地表水中ng/L级不同极性的农药及转化产物的同时测定。
2 实验部分
2.1 试剂与材料
莎稗磷标样(Dr. Ehrenstorfer GmbH公司):H同位素替代化合物\[6H2\]acephate(美国Cambridge Isotope Laboratories公司);其它单标标样(美国AccuStandard 公司)。甲醇、二氯甲烷、乙腈、乙酸乙酯(HPLC级,加拿大Fisher Scientific公司);甲酸铵(>99%)、乙酸铵(>99%)、甲酸(>99%)均购自美国SigmaAldrich公司。实验用水均为超纯水(美国Millipore 公司)。0.7 μm玻璃纤维滤膜及抽滤装置(美国Millipore公司),PSF GHP膜针头式过滤器(13 mm×0.2 μm,美国Pall公司),Visiprep DL固相萃取装置(美国Supelo公司)。Oasis HLB固相萃取小柱(500 mg,6 mL)、活性炭柱(SepPak Plus AC2 Cartridges, 400 mg)购自美国Waters公司;StrataX柱(500 mg, 6 mL)购自Phenomenex公司。
2.2 农药标准溶液及缓冲溶液的配制
100.0 mg/L 35种农药单标储备溶液,溶剂为甲醇; 2000 μg/L 35种农药的混合标准储备液,溶剂为甲醇; 0.5~200 μg/L混合标准工作溶液(均含10 μg/L Acephated6),溶剂为10 mmol/L乙酸铵的乙腈水(1∶1, V/V)混合溶液; 甲酸铵缓冲溶液(5 mmol/L,pH 3.5):用500 mL水溶解157 mg 甲酸铵,用约200 μL甲酸调至pH 3.5。
2.3 样品处理
水样采集至棕色玻璃瓶内,水样装满,瓶中不能留有空气。玻璃瓶在采集前用铬酸洗液润洗,以水冲洗。样品在4℃下避光保存。
2.4 固相萃取条件
取1.0 L水样,通过0.7 μm 玻璃纤维滤膜。上样至刚活化好的串联固相萃取小柱上(10 mL乙腈、10 mL甲醇和10 mL水活化),流速为5~10 mL/min。上样完成后,用10 mL水淋洗固相萃取柱,抽干30 min。两柱拆分,用10 mL乙腈洗脱HLB柱,用10 mL含20 mmol/L乙酸铵的甲醇洗脱活性炭柱,洗脱溶液混合后,40℃水浴氮吹,溶剂转化为含10 mmol/L乙酸铵的乙腈水(1∶1, V/V)混合溶液,定容至1.0 mL。在分析之前使用0.2 μm PSF GHP膜过滤。
2.5 UPLCMS/MS条件
实验使用ACQUITY UPLC/Quattro Premier XE液质联用仪、ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm, Waters 公司)。超高效液相色谱流动相A为5 mmol/L甲酸铵甲酸缓冲溶液(pH 3.5),流动相B为乙腈。梯度洗脱: 0~0.5 min, 5% B; 0.5~4.0 min, 5%~25% B; 4.0~8.0 min, 25%~32% B; 8.0~11.0 min, 32%~60% B; 11.0~18.5 min, 60%~100% B; 18.5~19.0 min, 100% B。流速:0.2 mL/min,进样体积:10 μL; 色谱柱温度:30℃; 质谱参数见表1。
3 结果与讨论
3. 1 前处理条件优化
3.1.1 固相萃取柱的选择 已有研究表明对水中的强极性有机磷农药乙酰甲胺磷、甲胺磷和氧化乐果Symbolm@@ 0.85≤lgKow≤Symbolm@@ 0.74),聚合柱SDB柱和HLB柱的富集效率分别为100%~15%和0%[24],活性炭15,15[BHDFG6*2,WKZQ0W]其它质谱条件:毛细管电压, 3.5 kV; 离子源温度, 120℃; 脱溶剂气氮气, 380℃; 脱溶剂气流速,600 L/h; 载气流速(50 L/h); 碰撞气氩气流速,0.15 mL/min.“*”:定量离子。
The other parameters:Capillary voltage, 3.5 kV; Source temperature, 120℃; Desolvation gas (N2) temperature, 380℃; Cone gas flow rate,600 L/h; Carrier gas flow rate 50 L/h; Collision gas argon flow rate, 0.15 mL/min.“*”: Quantitative ion pair.
柱的富集效率为76.4%~96.4%[28],但聚合柱SDB和HLB对水中不同极性农药(0.57≤lgKow≤4.96)富集效率范围分别为2.7%~260.6%和43.7%~115.3%[18]。为实现多极性农药Symbolm@@ 0.85≤lgKow≤5.2)的同时富集,本实验选择聚合柱和活性炭柱串联作优化实验。选取典型强极性有机磷农药乙酰甲胺磷(2号)代表强极性有机磷农药(1, 2, 3, 6, 7, 8, 9号),与其它类别农药(4, 5, 10~35号农药)同时作为目标化合物进行优化实验。
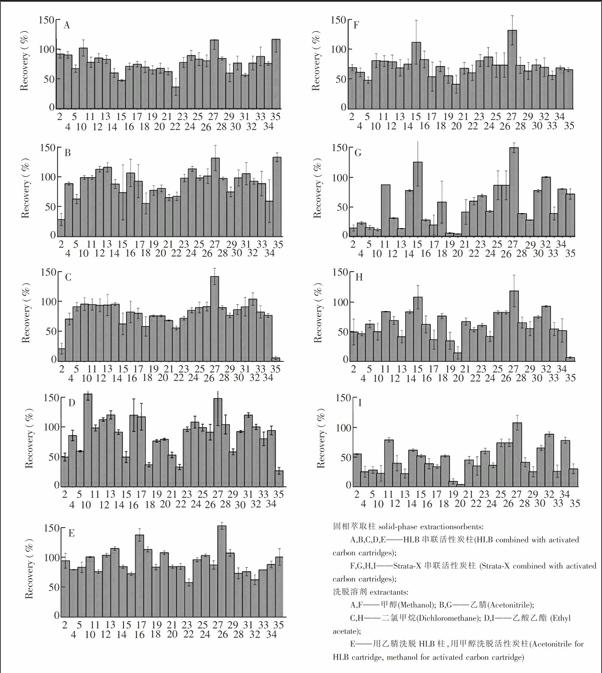
比较了聚合柱HLB柱和StrataX柱分别串联活性炭柱(聚合柱在上,活性炭柱在下)对目标物的回收率(图1)。不同洗脱溶剂下,用HLB柱串联活性炭柱,目标物的平均回收率范围为37%~117%(图1A甲醇)、29%~133%(图1B乙腈)、21%~142%(图1C二氯甲烷)、27%~155%(图1D乙酸乙酯)。用StrataX串联活性炭柱,目标物的平均回收率范围为:42%~132%(图1F甲醇)、4%~149%(图1G乙腈)、7%~118%(图1H二氯甲烷)、9%~108%(图1I乙酸乙酯)。在StrataX串联活性炭柱下,硫双威(17号)、扑灭津(19号)、特丁津(20号)和二嗪农(29号)的回收率较低。为保证更多目标物的萃取效率,最终选择了HLB串联活性炭柱。
基质, 超纯水; 水样体积, 500 mL;洗脱溶液体积, 10 mL;加标量, 20 ng/L。图中各种农药的编号同表1。
Matrix, ultrapure water; Water volume, 500 mL; Elution solvent volume, 10 mL; Amount added: 20 ng/L. The number of pesticide is the same as in Tabel 1。
3.1.2 洗脱溶剂的选择
考察了不同洗脱溶剂对29种农药(2,4,5,10~35)回收率的影响(图1):甲醇、乙腈、二氯甲烷、乙酸乙酯、乙腈和甲醇分别洗脱两柱。结果显示,甲醇、乙腈、二氯甲烷对大多数农药的洗脱效果较好(图1A,图1B,图1C和图1E)。特别地,甲醇能从活性炭柱上洗脱乙酰甲胺磷(2号),而乙腈、乙酸乙酯、二氯甲烷的洗脱效果较差[25];甲醇和乙腈能较好地洗脱灭草松(35号),而二氯甲烷和乙酸乙酯对灭草松的洗脱效果差。考虑到应用到实际水样时,使用甲醇溶剂能使萃取液中的颜色呈现黄色[31],最终选用乙腈作为HLB柱的洗脱溶剂。
为进一步优化对强极性化合物的的萃取效率,考察了4种4溶剂(甲醇、10 mmol/L乙酸铵甲醇、20 mmol/L乙酸铵甲醇、40 mmol/L乙酸铵甲醇)对7种强极性有机磷农药(甲胺磷(1号)、乙酰甲胺磷(2号)、氧化乐果(3号)、亚砜磷(6号)、久效磷(7号)、黄草灵(8号)、蚜灭多(9号))回收率的影响(图2)。采用串联固相萃取柱,HLB柱用乙腈洗脱,活性炭柱用以上4种溶剂洗脱。结果表明,洗脱溶剂为纯甲醇时,氧化乐果和亚砜磷的回收率在16%~37%之间;甲醇中加入乙酸铵,能够明显提高两者的回收率(89%~103%)。比较不同浓度的乙酸铵,含有20 mmol/L乙酸铵得到回收率相对标准偏差较小(1%≤RSD≤8%)。故本实验最终选择用10 mL 20 mmol/L乙酸铵甲醇洗脱活性炭柱。
综合考虑,最终选择了用10 mL乙腈为HLB柱洗脱溶剂,用10 mL 20 mmol/L乙酸铵甲醇为活性炭柱的洗脱溶剂。
3.2 色谱和质谱条件优化
分别将1 mg/L的单标标准溶液通过质谱直接进样,用于化合物质谱条件优化。灭草松采用ESI模式,其他化合物采用ESI+模式。选择各准分子离子[M+H]+或[M-H]
摘 要 采用固相萃取(SPE)超高效液相色谱串联质谱(UPLCMS/MS)技术,建立了同时测定环境水样中痕量浓度多极性的35种高关注农药及转化产物的方法。优化的前处理方法为:串联HLB和活性炭小柱用于富集水样,10 mL乙腈为HLB柱洗脱溶剂,10 mL 20 mmol/L乙酸铵甲醇作为活性炭柱的洗脱溶剂,将洗脱溶液混合、浓缩、溶剂转化和定容,定容溶剂为10 mmol/L乙酸铵的乙腈水的混合溶液(1∶1, V/V)。流动相分别为5 mmol/L甲酸铵甲酸缓冲溶液(pH 3.5)和乙腈进行UPLC色谱分离,质谱用多反应监测(MRM)模式进行分析测定。实验结果表明,目标物在0.5~200 μg/L含量范围内线性良好(R2≥0.9802),检出限范围为0.01~0.8 ng/L,回收率在60%~120%之间。本研究构建的SPEUPLCMS/MS方法灵敏可靠,可用于对地表水中ng/L级极性不同的多种农药及转化产物的测定。
关键词 农药; 转化产物; 固相萃取; 超高效液相色谱串联质谱法; 地表水
1 引 言
农药的大量使用造成各种环境基质中存在农药残留。农药对地表水形成污染的主要原因在于大部分残留在土壤或漂浮于大气中的农药通过雨水沉降和地表径流的方式进入地表水。农药在环境中通过水解、氧化、生物降解和光解作用形成转化产物[1]。转化产物具有比母体化合物更强的极性和水溶性,其在地表水高浓度检出和潜在的健康风险引起人们广泛关注[1~3]。部分农药及转化产物已被美国环保局(US EPA)列入饮用水污染物候选清单中(CCL3)中,如乙酰甲胺磷、甲胺磷、乐果、灭多威、异丙威、特丁磷砜、戊唑醇、草达灭、虫酰肼和乙草胺等。目前,国内针对地表水中的农药污染调查尚不系统和全面[4]。6种高毒有机磷品种(敌敌畏、甲基对硫磷、对硫磷、氧化乐果、甲胺磷、久效磷)的产量占国内有机磷杀虫剂产量的70%,占农药总产量的33%[5],但《国内地表水环境质量标准(GB38382002)》中仅规定了前3种有机磷农药(敌敌畏、对硫磷、甲基对硫磷)的标准限值,尚未对后3种强极性有机磷农药做出限值规定。研究表明,甲胺磷、氧化乐果等有机磷农药具有强极性特征,其辛醇水分配系数小于零,广泛存在于地表水中[6]。
农药种类繁多,各类农药的极性差别较大,因而分析方法也显著不同。针对环境水样中的有机磷农药或转化产物,分析方法主要为气相色谱法,检测器为氢火焰离子化检测器[7]或火焰光度检测器[6]。气相色谱串联质谱法(GCMS/MS)[8~10]在测定有机磷转化产物时也被使用,但在测定前需衍生化。液相色谱质谱法(LCMS)[11]或液相色谱串联质谱法(LCMS/MS)[12~14]可用于检测分析环境水样中多类别农药或转化产物。与GCMS相比,LCMS/MS方法在测定农药种类范围及检测灵敏度方面更具优势[15]。与固相微萃取法(SPME)[16]和液相微萃取法(LPME)[17]相比,固相萃取法(SPE)有较高的富集倍数,是目前富集水样中农药转化产物主要的前处理方法[18]。富集农药及转化产物的商业化和通用性的SPE小柱主要有C18柱[11,19],聚合柱[20,21]和石墨化炭黑柱[22]。聚合柱因为同时含有亲水基团和疏水基团,对多数农药有较好的富集效果[21,23],但是对于极性特强的有机磷农药(如乙酰甲胺磷、甲胺磷、氧化乐果)的萃取效率低于15%[24,25]。活性炭柱已被应用于富集水中丙烯酰胺[26]、二甲基亚硝胺[27]等强极性化合物。Hayama等[28]利用活性炭柱固相萃取亲水作用色谱分离测定环境水样中的6种强极性有机磷农药,回收率在76.4%~98.6%之间。然而,活性炭柱具有较强的吸附能力,水体中的天然有机物[29]、痕量有机污染物[29]和重金属离子[30]均被富集在萃取小柱上,大量干扰物的存在可能会影响强极性有机磷农药在UPLCMS/MS上的色谱分离和质谱分析。
针对以上问题,本研究优化聚合柱,串联活性炭柱,减少杂质的吸附,同时提高极性较弱的农药的富集效率,建立一种同时测定环境水样中浓度在ng/L水平的极性跨度大(
Symbolm@@ 0.85≤lgKow≤5.2)的35种农药及转化产物的SPEUPLCMS/MS方法。本实验选择了国内产量较高的农药,特别是强极性有机磷农药、列入US EPA CCL3中的部分农药及转化产物,作为目标化合物。采用HLB柱和活性炭柱串联,可用于地表水中ng/L级不同极性的农药及转化产物的同时测定。
2 实验部分
2.1 试剂与材料
莎稗磷标样(Dr. Ehrenstorfer GmbH公司):H同位素替代化合物[6H2]acephate(美国Cambridge Isotope Laboratories公司);其它单标标样(美国AccuStandard 公司)。甲醇、二氯甲烷、乙腈、乙酸乙酯(HPLC级,加拿大Fisher Scientific公司);甲酸铵(>99%)、乙酸铵(>99%)、甲酸(>99%)均购自美国SigmaAldrich公司。实验用水均为超纯水(美国Millipore 公司)。0.7 μm玻璃纤维滤膜及抽滤装置(美国Millipore公司),PSF GHP膜针头式过滤器(13 mm×0.2 μm,美国Pall公司),Visiprep DL固相萃取装置(美国Supelo公司)。Oasis HLB固相萃取小柱(500 mg,6 mL)、活性炭柱(SepPak Plus AC2 Cartridges, 400 mg)购自美国Waters公司;StrataX柱(500 mg, 6 mL)购自Phenomenex公司。
2.2 农药标准溶液及缓冲溶液的配制
100.0 mg/L 35种农药单标储备溶液,溶剂为甲醇; 2000 μg/L 35种农药的混合标准储备液,溶剂为甲醇; 0.5~200 μg/L混合标准工作溶液(均含10 μg/L Acephated6),溶剂为10 mmol/L乙酸铵的乙腈水(1∶1, V/V)混合溶液; 甲酸铵缓冲溶液(5 mmol/L,pH 3.5):用500 mL水溶解157 mg 甲酸铵,用约200 μL甲酸调至pH 3.5。
2.3 样品处理
水样采集至棕色玻璃瓶内,水样装满,瓶中不能留有空气。玻璃瓶在采集前用铬酸洗液润洗,以水冲洗。样品在4℃下避光保存。
2.4 固相萃取条件
取1.0 L水样,通过0.7 μm 玻璃纤维滤膜。上样至刚活化好的串联固相萃取小柱上(10 mL乙腈、10 mL甲醇和10 mL水活化),流速为5~10 mL/min。上样完成后,用10 mL水淋洗固相萃取柱,抽干30 min。两柱拆分,用10 mL乙腈洗脱HLB柱,用10 mL含20 mmol/L乙酸铵的甲醇洗脱活性炭柱,洗脱溶液混合后,40℃水浴氮吹,溶剂转化为含10 mmol/L乙酸铵的乙腈水(1∶1, V/V)混合溶液,定容至1.0 mL。在分析之前使用0.2 μm PSF GHP膜过滤。
2.5 UPLCMS/MS条件
实验使用ACQUITY UPLC/Quattro Premier XE液质联用仪、ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm, Waters 公司)。超高效液相色谱流动相A为5 mmol/L甲酸铵甲酸缓冲溶液(pH 3.5),流动相B为乙腈。梯度洗脱: 0~0.5 min, 5% B; 0.5~4.0 min, 5%~25% B; 4.0~8.0 min, 25%~32% B; 8.0~11.0 min, 32%~60% B; 11.0~18.5 min, 60%~100% B; 18.5~19.0 min, 100% B。流速:0.2 mL/min,进样体积:10 μL; 色谱柱温度:30℃; 质谱参数见表1。
3 结果与讨论
3. 1 前处理条件优化
3.1.1 固相萃取柱的选择 已有研究表明对水中的强极性有机磷农药乙酰甲胺磷、甲胺磷和氧化乐果Symbolm@@ 0.85≤lgKow≤Symbolm@@ 0.74),聚合柱SDB柱和HLB柱的富集效率分别为100%~15%和0%[24],活性炭15,15[BHDFG6*2,WKZQ0W]其它质谱条件:毛细管电压, 3.5 kV; 离子源温度, 120℃; 脱溶剂气氮气, 380℃; 脱溶剂气流速,600 L/h; 载气流速(50 L/h); 碰撞气氩气流速,0.15 mL/min.“*”:定量离子。
The other parameters:Capillary voltage, 3.5 kV; Source temperature, 120℃; Desolvation gas (N2) temperature, 380℃; Cone gas flow rate,600 L/h; Carrier gas flow rate 50 L/h; Collision gas argon flow rate, 0.15 mL/min.“*”: Quantitative ion pair.
柱的富集效率为76.4%~96.4%[28],但聚合柱SDB和HLB对水中不同极性农药(0.57≤lgKow≤4.96)富集效率范围分别为2.7%~260.6%和43.7%~115.3%[18]。为实现多极性农药(
Symbolm@@ 0.85≤lgKow≤5.2)的同时富集,本实验选择聚合柱和活性炭柱串联作优化实验。选取典型强极性有机磷农药乙酰甲胺磷(2号)代表强极性有机磷农药(1, 2, 3, 6, 7, 8, 9号),与其它类别农药(4, 5, 10~35号农药)同时作为目标化合物进行优化实验。
比较了聚合柱HLB柱和StrataX柱分别串联活性炭柱(聚合柱在上,活性炭柱在下)对目标物的回收率(图1)。不同洗脱溶剂下,用HLB柱串联活性炭柱,目标物的平均回收率范围为37%~117%(图1A甲醇)、29%~133%(图1B乙腈)、21%~142%(图1C二氯甲烷)、27%~155%(图1D乙酸乙酯)。用StrataX串联活性炭柱,目标物的平均回收率范围为:42%~132%(图1F甲醇)、4%~149%(图1G乙腈)、7%~118%(图1H二氯甲烷)、9%~108%(图1I乙酸乙酯)。在StrataX串联活性炭柱下,硫双威(17号)、扑灭津(19号)、特丁津(20号)和二嗪农(29号)的回收率较低。为保证更多目标物的萃取效率,最终选择了HLB串联活性炭柱。
基质, 超纯水; 水样体积, 500 mL;洗脱溶液体积, 10 mL;加标量, 20 ng/L。图中各种农药的编号同表1。
Matrix, ultrapure water; Water volume, 500 mL; Elution solvent volume, 10 mL; Amount added: 20 ng/L. The number of pesticide is the same as in Tabel 1。
3.1.2 洗脱溶剂的选择
考察了不同洗脱溶剂对29种农药(2,4,5,10~35)回收率的影响(图1):甲醇、乙腈、二氯甲烷、乙酸乙酯、乙腈和甲醇分别洗脱两柱。结果显示,甲醇、乙腈、二氯甲烷对大多数农药的洗脱效果较好(图1A,图1B,图1C和图1E)。特别地,甲醇能从活性炭柱上洗脱乙酰甲胺磷(2号),而乙腈、乙酸乙酯、二氯甲烷的洗脱效果较差[25];甲醇和乙腈能较好地洗脱灭草松(35号),而二氯甲烷和乙酸乙酯对灭草松的洗脱效果差。考虑到应用到实际水样时,使用甲醇溶剂能使萃取液中的颜色呈现黄色[31],最终选用乙腈作为HLB柱的洗脱溶剂。
为进一步优化对强极性化合物的的萃取效率,考察了4种4溶剂(甲醇、10 mmol/L乙酸铵甲醇、20 mmol/L乙酸铵甲醇、40 mmol/L乙酸铵甲醇)对7种强极性有机磷农药(甲胺磷(1号)、乙酰甲胺磷(2号)、氧化乐果(3号)、亚砜磷(6号)、久效磷(7号)、黄草灵(8号)、蚜灭多(9号))回收率的影响(图2)。采用串联固相萃取柱,HLB柱用乙腈洗脱,活性炭柱用以上4种溶剂洗脱。结果表明,洗脱溶剂为纯甲醇时,氧化乐果和亚砜磷的回收率在16%~37%之间;甲醇中加入乙酸铵,能够明显提高两者的回收率(89%~103%)。比较不同浓度的乙酸铵,含有20 mmol/L乙酸铵得到回收率相对标准偏差较小(1%≤RSD≤8%)。故本实验最终选择用10 mL 20 mmol/L乙酸铵甲醇洗脱活性炭柱。
综合考虑,最终选择了用10 mL乙腈为HLB柱洗脱溶剂,用10 mL 20 mmol/L乙酸铵甲醇为活性炭柱的洗脱溶剂。
3.2 色谱和质谱条件优化
分别将1 mg/L的单标标准溶液通过质谱直接进样,用于化合物质谱条件优化。灭草松采用ESI模式,其他化合物采用ESI+模式。选择各准分子离子[M+H]+或[M-H]
Symbolm@@ 作为母离子,进行二级质谱优化。优化的目的是保证多反应监测模式下被测离子对在质谱中有较高的响应值。优化了母离子、子离子、锥孔和碰撞电压等重要的质谱参数,优化结果见表1。
选择了普遍适用的色谱柱BEH C18柱。为最大限度的保留强极性化合物,起始流动相比为设置为95∶5(V/V),标准溶液的总离子流图如图3所示。
3.3 线性关系、精密度和回收率
采用外标法进行定量分析,绘制的标准曲线在0.5~200 μg/L范围内呈良好的线性关系,相关系数R2≥0.9802。方法的检出限(LODs,S/N=3)和定量限(LOQs,S/N=10)范围分别为0.01~0.8 ng/L和0.02~2.0 ng/L(表2)。针对极性较弱的农药,检出限低于或接近于用GCMS[31~35]或GCMS/MS[36]技术测定得到的检出限。
向空白水中添加一定浓度的混合标准溶液,重复5次。按优化的实验条件分析,计算相对标准偏差(见表2),结果在2.0%~22.7%之间。
[KH*4D][HT5”SS][HJ*4]表2 35种农药及转化产物的线性关系,线性范围,基质效应,检出限
3.4 基质效应评价
将地表水水样进行富集得到浓缩液,向浓缩液中加入混合标准溶液(浓度为10 μg/L),考察基质效应(Matrix effect,ME)。基质效应按公式(1)计算:
ME(%)=[1-(Rs+x-Rx)/Rs]×100(1)
式中,Rs+x和Rx分别为加入和未加入混标溶液的浓缩液中目标物的峰面积; Rs表示相同浓度水平标准样品中农药及转化产物的峰面积。不同目标物表现出不同程度的基质效应。总体上,大多数目标物的基质效应在30%以下(表2),特别是强极性有机磷农药(1, 2, 3, 6, 7, 8, 9号)。在活性炭柱前串联HLB柱,使得极性较强的有机磷农药与杂质共洗脱的可能性降低。
3.5 水源地的地表水样品分析
采用本方法对被用作水源地的地表水样品进行分析(表3)。检出的农药种类主要有甲胺磷、啶虫咪、乙草胺、戊唑醇、异丙威、敌敌畏等农药或转化产物,浓度范围在低ng/L级至ng/L级,确证了本方法能够测定地表水中低ng/L浓度水平下的农药或转化产物。
将标准溶液分别加入上述3个平行水样(加入体积100 μL),经过优化的固相萃取方法处理,计算回收率(表3),大多数农药或转化产物的回收率在60%~120%之间。草达灭和敌敌畏沸点较低(分别为259℃和251℃),在25℃下蒸气压分别为0.0056和0.0158 mm Hg,在氮吹过程中易挥发损失,导致这两种农药或转化产物的回收率并不高(<60%)[37]。实验结果表明,本方法灵敏可靠,可用于对地表水中ng/L级不同极性的农药及转化产物的同时测定。
References
1 Fenner K, Canonica S, Wackett L P, Elsner M. Science, 2013, 341(6147): 752-758
2 Richardson S D, Ternes T A. Anal. Chem., 2014, 86(6): 2813-2848
3 Olsson O, Khodorkovsky M, Gassmann M, Friedler E, Schneider M, Dubowski Y. CLEANSoil, Air, Water, 2013, 41(2): 134-142
4 SONG NingHui, BU YuanQing, SHAN ZhengJun. Journal of Ecology and Rural Environment, 2010, (S1): 49-57
宋宁慧, 卜元卿, 单正军. 生态与农村环境学报, 2010, (S1): 49-57
5 WANG lvXian. Modern Chemical Industry, 2000, 20(2): 1-6
王律先. 现代化工, 2000, 20(2): 1-6
6 Zhang Z L, Hong H S, Zhou J L, Yu G. J. Environ. Monit., 2002, 4(4): 498-504
7 Bavcon M, Trebse P, ZupancicKralj L. Chemosphere, 2003, 50(5): 595-601
8 HUANG XiaoHui, XUE Jian, XU XiaoLong, LIU Yan, ZHANG Jian. Journal of Analytical Science, 2013, 29(3): 409-412
黄晓会, 薛 健, 徐小龙, 刘 艳, 张 健. 分析科学学报, 2013, 29(3): 409-412
9 De Alwis G K H, Needham L L, Barr D B. J. Chromatogr. B, 2006, 843(1): 34-41
10 HUANG BaoYong, PAN CanPing, WANG YiRu, CAO Zhe, ZHANG Wei, LI WenMing, JIANG ShuRen. Chem. J. Chinese Universities, 2006, 27(2): 227-232
黄宝勇, 潘灿平, 王一茹, 曹 喆, 张 微, 李文明, 江树人. 高等学校化学学报, 2006, 27(2): 227-232
11 Ferrer I, Thurman E M. J. Chromatogr. A, 2007, 1175(1): 24-37
12 Reemtsma T, Alder L, Banasiak U. J. Chromatogr. A, 2013, 1271(1): 95-104
13 YAN Rui,SHAO MingYuan,JU FuLong,SONG DaQian,ZHANG HanQi,YU AiMin. Chinese J. Anal.Chem., 2013, 41(2): 315-316
闫 蕊, 邵明媛, 鞠福龙, 宋大千, 张寒琦, 于爱民. 分析化学, 2013, 41(2): 315-316
14 Greulich K, Alder L. Anal. Bioanal. Chem., 2008, 391(1): 183-197
15 Alder L, Greulich K, Kempe G, Vieth B. Mass Spectrom. Rev., 2006, 25(6): 838-865
16 Bagheri H, Ayazi Z, Babanezhad E. Microchem. J., 2010, 94(1): 1-6
17 Lambropoulou D A, Albanis T A. J. Biochem. Bioph. Methods, 2007, 70(2): 195-228
18 Vidal J L M, PlazaBolanos P, RomeroGonzalez R, Frenich A G. J. Chromatogr. A, 2009, 1216(40): 6767-6788
19 Yokley R A, Mayer L C, Huang S B, Vargo J D. Anal. Chem., 2002, 74(15): 3754-3759
20 Benvenuto F, Marin J M, Sancho J V, Canobbio S, Mezzanotte V, Hernandez F. Anal. Bioanal. Chem., 2010, 397(7): 2791-2805
21 Rodrigues A M, Ferreira V, Cardoso V V, Ferreira E, Benoliel M J. J. Chromatogr. A, 2007, 1150(12): 267-278
22 Shoemaker J A, Bassett M V. J. AOAC Int., 2006, 89(1): 201-209
23 Primel E G, Caldas S S, Escarrone A L V. Cent. Eur. J. Chem., 2012, 10(3): 876-899
24 Ingelse B A, van Dam R C J, Vreeken R J, Mol H G J, Steijger O M. J. Chromatogr. A, 2001, 918(1): 67-78
25 Liu F M, Bischoff G, Pestemer W, Xu W, Kofoet A. Chromatographia, 2006, 63(56): 233-237
26 CHEN Ling, CHEN Hao. Chinese Journal of Chromatography, 2003, 21(5): 534
陈 玲, 陈 皓. 色谱, 2003, 21(5): 534
27 LIANG Chuang, XU Bin, XIA ShengJi, GAO NaiYun, LI DaPeng, TIAN FuXiang. China Water & Wastewater, 2009, 25(14): 82-85, 92
梁 闯, 徐 斌, 夏圣骥, 高乃云, 李大鹏, 田富箱. 中国给水排水, 2009, 25(14): 82-85, 92
28 Hayama T, Yoshida H, Todoroki K, Nohta H, Yamaguchi M. Rapid Commun. Mass Spectrom., 2008, 22(14): 2203-2210
29 Quinlivan P A, Li L, Knappe D R U. Water Res., 2005, 39(8): 1663-1673
30 ZHOU QiaoLi, LEI YongQian, GUO PengRan, PAN JiaChuan, WANG GuanHua. Journal of Instrumental Analysis, 2015, 34(2): 221-226
周巧丽, 雷永乾, 郭鹏然, 潘佳钏, 王冠华. 分析测试学报, 2015, 34(2): 221-226
31 Chang H, Wan Y, Hu J. Environ. Sci. Technol., 2009, 43(20): 7691-7698
32 Lindley C E, Stewart J T, Sandstrom M W. J. AOAC Int., 1996, 79(4): 962-966
33 Nakamura S, Daishima S. Anal. Bioanal. Chem., 2005, 382(1): 99-107
34 Tavakoli M, Hajimahmoodi M, Shemirani F. Anal. Methods, 2014, 6(9): 2988-2997
35 Yurtkuran Z, Saygi Y. Bull. Environ. Contam. Toxicol., 2013, 91(2): 165-170
36 Penetra A, Cardoso V V, Ferreira E, Benoliel M J. Water Sci. Technol., 2010, 62(3): 667-675
37 KockSchulmeyer M, Ginebreda A, Postigo C, Garrido T, Fraile J, de Alda M L, Barcelo D. Sci. Total Environ., 2014, 470: 1087-1098
Abstract A method based on solid phase extraction combined with ultraperformance liquid chromatographytandem mass spectrometry (SPEUPLCMS/MS) was established for the simultaneous determination of thirtyfive pesticides and transformation products of high concern with different polarities at trace level in environmental waters. Under optimized conditions, tandem cartridges with Oasis HLB column and activated carbon were used for the enrichment of water samples. HLB column was eluted by 10 mL of acetonitrile, and activated carbon cartridge was eluted by 10 mL of methanol solution containing 20 mmol/L ammonium acetate. The elution solvents were mixed, concentrated and reconstituted. Finally, the elution solvent was reconstituted with a mixed solution of acetonitrile and water (1∶1, V/V) containing 10 mmol/L ammonium acetate. The targets were separated by gradient elution with 5 mmol/L ammonium formate buffer and acetonitrile as mobile phases, and detected by MS/MS in the multireactions monitoring (MRM) mode. Under the experimental conditions, good linearity ranging from 0.5-200 μg/L was made (R2≥0.9802), and the limits of detection (LODs) were from 0.01 ng/L to 0.8 ng/L. Recoveries of most targets were 60%-120%. The SPEUPLCMS/MS method established here is reliable and can be applied to determine ng/L level of pesticides and transformation products with different polarities in surface water.
