高效液相色谱—三重四级杆/复合线性离子阱质谱测定鼠脑组织中神经甾体类化合物
2015-08-13刘佳等
刘佳等

摘 要 建立了高效液相色谱串联质谱法(HPLCMS/MS)测定鼠脑组织中6种神经甾体类激素(别孕烯醇酮、孕烯醇酮、孕酮、脱氧皮质酮、四氢脱氧皮质酮、去氢表雄酮)的方法。采用Agilent XDBC18色谱柱,以5 mmol/L乙酸铵甲醇为流动相,梯度洗脱分离,在电喷雾正离子模式下,以多反应监测(MRM)扫描模式采集数据,基质匹配内标法定量。孕酮和脱氧皮质酮在0.10~10 ng/mL范围内呈良好的线性关系,别孕烯醇酮、孕烯醇酮、四氢脱氧皮质酮、去氢表雄酮在0.50~10 ng/mL范围内呈良好的线性关系,相关系数均大于0.99; 在0.4, 4.0和8.0 ng/mL加标水平下的平均回收率为91.2%~115.5%;相对标准偏差(RSD,n=6)为0.87%~8.8%;检出限为0.04~0.20 ng/mL,定量限为0.10~0.50 ng/mL。
关键词 神经甾体; 液相串联质谱; 多重反应监测; 鼠脑
1 引 言
神经甾体是指在神经系统中合成的甾体和经血脑屏障进入神经系统发挥作用的外周甾体及其代谢衍生物[1]。这些神经甾体可以通过结合不同的神经受体(如GABAA,Sigma受体)调控神经功能。神经甾体的行为与中枢神经系统正常功能的行使密切相关。研究表明, 在不同的生理和病理条件下,神经甾体在各个脑区中的含量会显著变化[2,3]。神经甾体还会参与焦虑和抑郁的调节过程。由于神经甾体在生物体内含量极低,基质干扰多,使得生物体内内源性神经甾体的测定十分困难,因而开发快速、准确检测生物体内神经甾体含量的方法就显得尤为重要。
传统的神经甾体的检测方法有放射免疫标记法(RIA)和气相色谱质谱联用法(GCMS)[4],但放射免疫标记法特异性差,且存在安全隐患;气相色谱质谱联用法虽然灵敏度高,但样品前处理步骤复杂,不能实现高通量分析。液相色谱质谱联用技术由于其选择性好、灵敏度高、适用范围广等优点,近年被广泛应用到神经甾体检测领域[5~8]。但这些工作有的需要复杂的衍生化过程[9],有的使用多个同位素内标,增加了检测成本[10];有的只检测1~2种待测物,缺乏全面性[7,11]。
本研究以大鼠脑组织为基质,建立了高效液相色谱串联质谱法(HPLCMS/MS)同时检测脑组织中别孕烯醇酮(AP)、孕烯醇酮 (PREG)、孕酮(PROG)、脱氧皮质酮(DOC)、四氢脱氧皮质酮(THDOC)、去氢表雄酮(HEA)6种神经甾体类化合物的方法。本实验采用的高效液相色谱三重四级杆/复合线性离子阱质谱是一种新型质谱仪,与普通的液相色谱三重四级杆串联质谱相比,提高了MS/MS扫描能力。本研究以甲基睾酮为内标,较同位素标记物内标方便易得,内源性杂质不干扰测定,提高了方法的准确度和精密度[12]。本方法无需衍生化,操作简便、快速,可同时测定大鼠不同脑区中神经甾体的含量,为建立其他生物样本中神经甾体的检测方法提供参考。
2 实验部分
2.1 仪器与试剂
LC20ADXR高效液相色谱(日本岛津公司);AB SCIEX QTRAP 6500质谱仪(美国AB公司);固相萃取仪(美国Supelco公司);固相萃取柱Strata C18E 50 mg/1 mL(美国菲罗门公司);离心浓缩仪(美国Thermo公司);0.22 μm聚丙烯(Polypropylene)膜过滤离心管(美国Corning公司);MilliQ超纯水仪(美国Millipore公司)
标准品别孕烯醇酮(Allopregnanolone,AP,纯度98%)、孕烯醇酮 (Pregnenolone,PREG,纯度98%)、孕酮(Progesterone,PROG,纯度99%)、脱氧皮质酮(11Deoxycorticosterone,DOC,纯度97%)、四氢脱氧皮质酮(Allotetrahydrodoc,THDOC,纯度99.5%)、甲基睾酮(Methyltestosterone,MT,纯度99%)、去氢表雄酮(Dehydroepiandrosterone,DHEA,纯度99%)均为美国Sigma公司试剂。
甲醇(色谱纯,美国Fisher试剂);乙酸铵(纯度99%,美国Sigma公司);甲酸(纯度88%,美国Fisher公司);实验用水为MilliQ超纯水(电阻率18.2 MΩ cm)。
2.2 溶液配制
将各标准品用甲醇配制成质量浓度为1 mg/mL的标准储备液,并根据需要用甲醇水(1∶1, V/V)稀释成适当质量浓度的混合标准溶液,
Symbolm@@ 20℃保存。
2.3 样品前处理
2.3.1 标准曲线的构建 配制5% BSA0.7% NaCl溶液,加入不同浓度的AP,PREG,DOC,THDOC,PROG,DHEA标准品,浓度范围为0.1 ~ 10 ng/mL,再加入相同浓度(0.3 pg/mL)的内标物MT,振荡混匀后进行液液萃取。加入1.2 mL乙酸乙酯正己烷(9∶1, V/V),涡旋5 min,于12000 r/min下离心5 min,将上层有机相转入另一洁净离心管中。氮气吹干,100 μL甲醇水(1∶1, V/V,0.1% 甲酸)复溶。12000 r/min离心15 min,上清液供HPLCMS/MS分析。
2.3.2 鼠脑组织 大鼠采用异氟烷麻醉,断头取血,冰浴分离内侧前额叶皮层(mPFC)、海马(HF)和丘脑腹后核(VP),液氮保存,称重(准确称至0.1 mg)。每份组织中加入0.49 mL超纯水,10 μL 15 pg/mL MT,电动匀浆机匀浆,超声。采用液液萃取法(见2.3.1节)和固相萃取法(见2.3.3节)两种方法处理。
2.3.3 固相萃取方法 预先以1 mL甲醇、1 mL水活化平衡Strata C18柱。将组织匀浆液于12000 r/min下离心5 min,然后将上清液加入Strata C18柱中; 以1 mL 5%甲醇作为淋洗液,淋洗Strata C18柱。抽至近干后,以1 mL甲醇洗脱。收集洗脱液,常温下用N2吹干。100 μL甲醇水(1∶1, V/V,0.1% 甲酸)复溶。12000 r/min离心15 min,上清液供UFLCMS/MS分析。
2.4 色谱条件
Agilent XDBC18色谱柱( 50 mm×4.6 mm,1.8 μm,美国安捷伦公司)。流动相:5 mmol/L乙酸铵溶液(A)和甲醇(B);采用梯度洗脱模式:0~2.5 min,60% B;2.5~5.5 min,60%~95% B;5.5~7.0 min,95% B;7.0~7.1 min,95%~60% B;7.1~10.0 min,60% B。流速0.5 mL/min。进样体积10 μL;样品室温度为室温;柱温45℃;采用内标法进行定量分析。
2.5 质谱条件
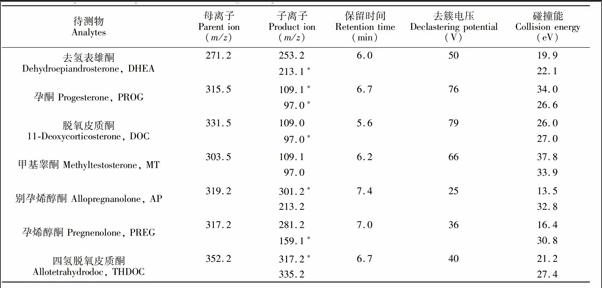
电喷雾离子源(ESI),正离子扫描方式,多反应监测扫描(MRM)模式。离子化电压(IS):5500 V,气帘气(CUR):241.33 kPa,喷雾气(GS1):413.7 kPa,辅助加热气(GS2):413.7 kPa,碰撞器(CAD): Medium。由于THDOC的氨加合物物受热容易分解,离子源温度(TEM)设为100℃。各物质离子对优化后参数见表1。
3 结果与讨论
3.1 质谱条件的建立
神经甾体类化合物既可以用电喷雾离子源(ESI)电离,也可以用大气压化学电离源(APCI)分析[10],本研究采用ESI正离子模式作为其离子化方式。取浓度为1.0 mg/L的各神经甾体标准溶液,采用微量蠕动泵连续进样,一级质谱全扫描,确定准分子离子。结果显示,6种神经甾体均在正离子模式下获得准分子离子峰。对除THDOC外, 其余5种神经甾体以母离子为准分子离子,对其进行二级质谱扫描,选丰度最高的两个特征碎片离子作为定性定量离子,并优化相应离子对的去簇电压(Declusteringpotential, DP)、碰撞能量(Collision energy, CE)、碰撞室出口电压(Collision cell exit potential, CXP)和
3.2 液相色谱条件的选择
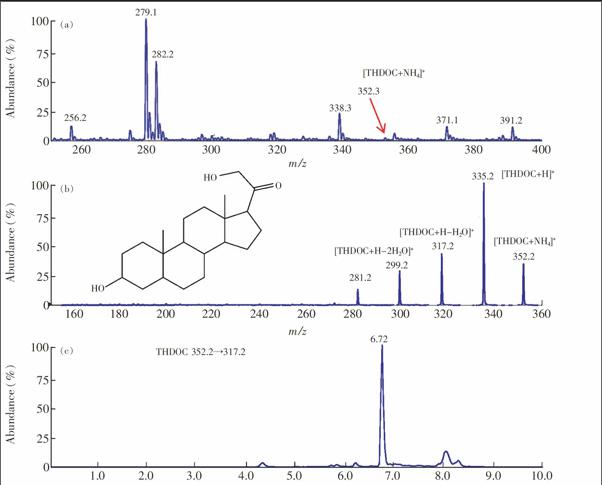
3.2.1 色谱柱对分离的影响 6种甾体类激素化学结构相似,如别孕烯醇酮(AP)与孕烯醇酮(PREG)分子结构十分相近,尽管MRM方式不要求所有峰都基线分离,但分离不佳易引起共流出物之间的互相抑制。本研究分别比较了Shimpack XRODS(50 mm × 4.6 mm,2.2 μm, 岛津公司)、Shimpack XRODS(100 mm×4.6 mm,2.2 μm, 岛津公司)、XDBC18(50 mm×4.6 mm,1.8 μm, 安捷伦公司)、Synergi Hydro RP(150 mm×2.0 mm,4 μm, 菲罗门公司)和Venusil XBP C18( 150 mm×2.1 mm,5 μm, 安吉尔公司)5种色谱柱对6种甾体类化合物的分离效果。结果表明, XDBC18色谱柱对6种甾体类化合物的分离效果最好, 因此本研究以其为分离柱。6种甾体类化合物的保留时间见表1,相应的结构式和MRM色谱图见图2。
3.2.2 流动相对分离的影响 以XDBC18为色谱柱,考察不同流动相对6种甾体类化合物分离的影响。分别对比了在A相和B相中加入酸或缓冲盐时的检测结果。结果表明,A相为纯水时对甾体类化合物色谱峰分离信号响应, 优于酸化纯水(0.1% 甲酸),可能因为甲酸改变了流动相的pH值,进而影响了待测物在分析柱上的保留。在水中添加挥发性缓冲盐乙酸铵,比较添加前后对甾体类物质的保留时间、分离度、色谱峰形、灵敏度等的影响情况。结果表明,A相为5 mmol/L乙酸铵溶液,B相为纯甲醇时,分离效果最佳。
3.2.3 柱温对的影响 考察了柱温箱温度为35℃和45℃时对信号的影响,样品是1 ng/mL混合标样。柱温在45℃时待测物保留时间会缩短。对于AP,当柱温为35℃时,其响应淹没于本底噪音中;当柱温为45℃时,可以看到明显的AP峰。由于神经甾体在组织中浓度很低,仪器灵敏度是优先考虑的问题,因此柱温选择为45℃。
3.3 生物样品前处理条件的优化
3.3.1 液液萃取(LLE)和固相萃取(SPE)的比较 液液萃取和固相萃取是生物样品常用的两种样品前处理技术,两者均有其各自的优势与特点。液液萃取简单、廉价,但背景干扰高于固相萃取。本研究以THDOC为例,比较了两种萃取方法对于提取鼠脑组织中的神经甾体的效果。结果表明,液液萃取的效果明显优于固相萃取。而且液液萃取1次后,THDOC的响应高于液液萃取3次,这可能是由于萃取的次数过多导致脑组织中的杂质也进入到萃取液中。图3展示了不同萃取条件下,海马组织中THDOC的响应情况。
3.3.2 内标物的选择 本研究为了体现方法的通用性和简便性,没有采用昂贵的同位素内标,而是选择甲基睾丸酮(MT)作为内标,其结构式见图2。MT较同位素内标更为方便易得,内源性杂质不干扰测定,内标与样品的提取回收率相近,提高了方法的精密度和准确度[12]。
3.4 方法验证
3.4.1 线性范围检出限定量限 由于存在基质效应,标准品在甲醇/水溶液中的质谱响应与其在生物基质中的响应不同,所以应将不同浓度的标准品加入到空白基质中,以制作工作曲线。而神经甾体属于内源性物质,无法得到空白基质,于是选用5%BSA0.7% NaCl作为空白基质[7]。配制浓度为0.1, 0.2, 0.5, 1, 2, 510 ng/mL的6种神经甾体类化合物的溶液,按2.3节的方法处理并测定,平行测定3次。以目标组分峰面积/内标峰面积为纵坐标,响应的添加含量为横坐标,绘制标准曲线。依据特征离子MRM色谱峰的信噪比大于3为检出限,信噪比大于10为定量限,结果见表2。6种神经甾体激素的LODs为0.04~0.20 ng/mL,定量限LOQs确定为0.10~0.50 ng/mL。DOC和PROG在0.1~10 ng/mL范围内呈良好的线性关系,PREG,THDOC,AP和DHEA在0.5~10 ng/mL范围内呈良好的线性关系。
[KH*4D][HT5”SS][HJ*4]表2 6种神经甾体类化合物的线性方程、相关系数、检出限和定量限
去氢表雄酮 DHEAy=0.0106x+0.007460.99770.50~10 0.20[]0.50
孕酮 PROGy=0.644x+0.03690.99870.10~10 0.04[]0.10
脱氧皮质酮 DOCy=0.402x+0.0004740.99470.10~10 0.04[]0.10
别孕烯醇酮 APy=0.365x+0.09350.99930.50~10 0.10[]0.50
孕烯醇酮 PREGy=0.0406x+0.006650.99100.50~10 0.10[]0.50
四氢脱氧皮质酮 THDOCy=0.236x-0.01390.99930.50~10 0.20[]0.50
[BHDFG1*2,WKZQ0W][BG)W][HT5][HJ] 以5% BSA0.7% NaCl作为空白基质, 考察方法的可靠性[14]。即在鼠脑组织中加入不同浓度的标准品,比较以鼠脑组织作为基质和溶液作为空白基质时工作曲线的斜率,发现两者基本一致。实验表明,以5%BSA0.7% NaCl溶液作为空白基质测定神经甾体是可靠的。
3.4.2 方法的回收率与精密度 在线性范围内选择3个添加水平,分别为0.4,4.0和8.0 ng/mL,按照2.3~2.5节方法平行测定6次,各化合物的平均回收率为91.2%~115.5%,RSD为0.9%~8.8%,具体数据见表3。
3.5 实际样品分析
利用本方法测定了成年大鼠脑组织不同分区中的6种神经甾体的浓度,结果见表4。
4 结 论
本研究建立了高效液相色谱串联质谱(HPLCMS/MS)测定鼠脑组织中6种神经甾体类激素的检测方法。在样品前处理过程中,发现乙酸乙酯正己烷萃取1次效果良好;优化了高效液相色谱串联质谱法同时检测多种内源性神经甾体类激素的仪器条件;采用内标法同时对6种激素进行定量分析,均获得了较好的效果。该方法样品前处理简单、灵敏度高、选择性好,有望广泛地应用于生物组织中痕量甾体类激素的检测。
References
1 Robel P, Baulieu E E. Trends Endocrinol. Metab., 1994, 5(1): 1-8
2 Luchetti S, Huitinga I, Swaab D F. Neuroscience, 2011, 191: 6-21
3 Reddy D S, Rogawski M A. J. Neurosci., 2002, 22(9): 3795-3805
4 Liere P, Akwa Y, WeillEngerer S, Eychenne B, Pianos A, Robel P, Sjovall J, Schumacher M, Baulieu E E. J. Chromatogr. B, 2000, 739(2): 301-312
5 Caruso D, Scurati S, Maschi O, de Angelis L, Roglio I, Giatti S, GarciaSegura L M, Melcangi R C. Neurochem. Int., 2008, 52(45): 560-568
6 Keevil B G. Best Pract. Res. Clin. Endoc. Metab., 2013, 27(5): 663-674
7 Rustichelli C, Pinetti D, Lucchi C, Ravazzini F, Puia G. J. Chromatogr. B, 2013, 930: 62-69
8 Wang Y Q, Karu K, Griffiths W J. Biochimie, 2007, 89(2): 182-191
9 Ahonen L, KeskiRahkonen P, Saarelainen T, Paviala J, Ketola R A, Auriola S, Poutanen M, Kostiainen R. Anal. Chim. Acta, 2012, 721: 115-121
10 Galuska C E, Hartmann M F, SanchezGuijo A, Bakhaus K, Geyer J, Schuler G, Zimmer K P, Wudy S A. Analyst, 2013, 138(13): 3792-3801
11 KeskiRahkonen P, Huhtinen K, Desai R, Harwood D T, Handelsman D J, Poutanen M, Auriola S. J. Mass Spectrom., 2013, 48(9): 1050-1058
12 REN JinMin, HOU YanNing. Acta Phamaceutica Sinica, 2005, 40(3): 262-266
任进民, 侯艳宁. 药学学报, 2005, 40(3): 262-266
13 Jantti S E, Tammimaki A, Raattamaa H, Piepponen P, Kostiainen R, Ketola R A. Anal. Chem., 2010, 82(8): 3168-3175
14 Minkler P E, Stoll M S K, Ingalls S T, Yang S M, Kerner J, Hoppel C L. Clin. Chem., 2008, 54(9): 1451-1462
act An analytical method based on high performance liquid chromatography tandem mass spectrometry was developed for the simultaneous determination of 6 neurosteroids in rat brain tissues. The neurosteroids were separated on an Agilent XDBC18 column (50 mm×4.6 mm, 1.8 μm), using methanol5 mmol/L ammonium acetate as the mobile phase. A gradient elution program with a cycle time of 10 min was used. The neurosteroids were detected by electrospray ionization mass spectrometry in positive mode with multiple reaction monitoring (MRM) scan mode. For 11deoxycorticosterone (DOC) and progesterone (PROG), the linear range of calibration curve was from 0.10 ng/mL to 10 ng/mL, and for allopregnanolone (AP), pregnenolone (PREG), dehydroepiandrosterone (DHEA) and allotetrahydrodoc (THDOC), the linear range calibration curve was from 0.50 ng/mL to 10 ng/mL, both with the correlation coefficient more than 0.99. The mean recoveries at spiked levels of 0.4, 4.0 and 8.0 ng/mL were in the range of 91.2%-115.5%, and the RSDs were 0.9%-8.8% (n=6). The limits of detection and quantification were 0.04-0.20 ng/mL and 0.10-0.50 ng/mL, respectively.endprint
